the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 31 Aug 2020
| 31 Aug 2020
Coalescence of bacterial groups originating from urban runoffs and artificial infiltration systems among aquifer microbiomes
Yannick Colin
Rayan Bouchali
Laurence Marjolet
Romain Marti
Florian Vautrin
Jérémy Voisin
Emilie Bourgeois
Veronica Rodriguez-Nava
Didier Blaha
Thierry Winiarski
Florian Mermillod-Blondin
Benoit Cournoyer
The invasion of aquifer microbial communities by aboveground microorganisms, a phenomenon known as community coalescence, is likely to be exacerbated in groundwaters fed by stormwater infiltration systems (SISs). Here, the incidence of this increased connectivity with upslope soils and impermeabilized surfaces was assessed through a meta-analysis of 16S rRNA gene libraries. Specifically, DNA sequences encoding 16S rRNA V5-V6 regions from free-living and attached aquifer bacteria (i.e., water and biofilm samples) were analysed upstream and downstream of a SIS and compared with those from bacterial communities from watershed runoffs and surface sediments from the SIS detention and infiltration basins. Significant bacterial transfers were inferred by the SourceTracker Bayesian approach, with 23 % to 57 % of the aquifer bacterial biofilms being composed of taxa from aboveground sediments and urban runoffs. Sediments from the detention basin were found more significant contributors of taxa involved in the buildup of these biofilms than soils from the infiltration basin. Inferred taxa among the coalesced biofilm community were predicted to be high in hydrocarbon degraders such as Sphingobium and Nocardia. The 16S rRNA-based bacterial community structure of the downstream-SIS aquifer waters showed lower coalescence with aboveground taxa (8 % to 38 %) than those of biofilms and higher numbers of taxa predicted to be involved in the N and S cycles. A DNA marker named tpm enabled the tracking of bacterial species from 24 genera including Pseudomonas, Aeromonas and Xanthomonas, among these communities. Several tpm sequence types were found to be shared between the aboveground and aquifer samples. Reads related to Pseudomonas were allocated to 50 species, of which 16 were found in the aquifer samples. Several of these aquifer species were found to be involved in denitrification but also hydrocarbon degradation (P. aeruginosa, P. putida and P. fluorescens). Some tpm sequence types allocated to P. umsongensis and P. chengduensis were found to be enriched among the tpm-harbouring bacteria, respectively, of the aquifer biofilms and waters. Reads related to Aeromonas were allocated to 11 species, but only those from A. caviae were recovered aboveground and in the aquifer samples. Some tpm sequence types of the X. axonopodis phytopathogen were recorded in higher proportions among the tpm-harbouring bacteria of the aquifer waters than in the aboveground samples. A significant coalescence of microbial communities from an urban watershed with those of an aquifer was thus observed, and recent aquifer biofilms were found to be significantly colonized by runoff-opportunistic taxa able to use urban C sources from aboveground compartments.
- Article
(4955 KB) - Full-text XML
-
Supplement
(764 KB) - BibTeX
- EndNote





Urbanization exerts multiple pressures on natural habitats and particularly on aquatic environments (Konrad and Booth, 2005; McGrane, 2016; Mejía and Moglen, 2009). The densification of urban areas, combined with the conversion of agricultural and natural lands into urban land use, led to the replacement of vegetation and open fields by impervious urban structures (i.e., roads, rooftops, sidewalks and parking lots) (Barnes et al., 2001). These impervious structures reduce the infiltration capacity of soils. They also exacerbate the speed and volume of stormwater runoff that favour soil erosion and flooding events and affect adversely natural groundwater recharge processes (Booth, 1991; Shuster et al., 2005). Due to these consequences, stormwater infiltration systems (SISs) or managed aquifer recharge (MAR) systems have been developed over the last decades and are gaining more interest in developed countries (Pitt et al., 1999). Such practices reduce direct stormwater discharges to surface waters and alleviate water shortages (Barba et al., 2019; Dillon et al., 2008; Marsalek and Chocat, 2002). However, stormwater represents a major source of nonpoint pollution, and its infiltration into the ground may have adverse ecological and sanitary impacts (Chong et al., 2013; Pitt et al., 1999; Vezzaro and Mikkelsen, 2012).
The vadose zone of a SIS can act as a natural filter capturing pollutants (hydrocarbons and heavy metals) and microorganisms washed off by runoffs (e.g., Murphy and Ginn, 2000; Tedoldi et al., 2016). Nevertheless, the effectiveness of a SIS in preventing the migration of contaminants towards aquifers is not always optimal (Borchardt et al., 2007; Lapworth et al., 2012; Arnaud et al., 2015; Voisin et al., 2018). The filtering properties of a SIS are influenced by various abiotic factors such as the nature of the media (rocks, sand and other soil elements), the physical properties (e.g., granulometry, hydrophobicity index and organization) and the runoff water flow velocity (Lassabatere et al., 2006; Winiarski et al., 2013). These constraints will impact not only the water transit time from the top layers to the aquifer but also the biological properties of these systems including the plant cover, root systems, worm population and composition of microbiota (Barba et al., 2019; Bedell et al., 2013; Crites, 1985; Pigneret et al., 2016). The thickness of the vadose zone was found to be one of the key parameters explaining chemical transfers such as phosphate and organic-carbon sources (Voisin et al., 2018). The situation is much less clear regarding the microbiological communities that flow through these systems (e.g., Barba et al., 2019; Voisin et al., 2018).
According to the concept of microbial community coalescence conceptualized by Tikhonov (2016) and adapted to riverine networks by Mansour et al. (2018), urban aquifers fed by a SIS should harbour microbiota reflecting the coalescence (community assemblages and selective sorting) of aboveground microbial communities with those of the aquifer. Indeed, during rain events, microbial communities will be resuspended through runoff-driven surface erosion processes, favouring detachment of microorganisms from plant litter, waste, soil and other particles (Mansour et al., 2018). These resuspended communities will merge and generate novel assemblages. The resulting community will initially match the relative contributions of the various subwatersheds to the overall microbiological complexity of the assemblages (Mansour et al., 2018). The prevailing ecological constraints among the downward systems will then gradually drive this coalescence towards the most fit community structures. These resulting communities might be highly efficient at degrading urban pollutants trapped among a SIS but could also disturb the ecological equilibria of the connected and more sensitive systems like those of deep aquifers as suggested by Voisin et al. (2018).
This study explores the impact of a SIS with a thick vadose zone (>10 m) on the coalescence of urban runoff microbial communities in a connected aquifer. Two hypotheses were tested (1) highly specialized taxa (often termed K-strategists; e.g., Vadstein et al., 2018) of an aquifer outcompete the intrusive community members of aboveground taxa, and (2) nutrient inputs from runoffs and pollutants drive changes among communities and favour environmental opportunists (often termed r-strategists; e.g., Vadstein et al., 2018). The targeted SIS is part of a long-term experimental site (http://www.graie.org/portail/dispositifsderecherche/othu/, last access: May 2020) that records both physicochemical and biological properties. This SIS is connected to the eastern aquifer of Lyon (France), which is fed by three low hydraulic conductivity corridors (10−5–10−8 m s−1) separated by moraine hills (Foulquier et al., 2010). The average vadose-zone thickness of the SIS is 15 m, and the delay between a rainfall event and the impact on the aquifer waters was estimated at 86±11 h (Voisin et al., 2018). A 16S rRNA gene metabarcoding dataset was assembled for this site to investigate bacterial community coalescence from the top compartments into the connected aquifer waters but also the biofilm communities developing on inert surfaces. This investigation was also built on the hypothesis that a less significant microbial community coalescence is expected in aquifer waters than biofilms. This is supported by previous reports which suggested the occurrence of transient free-living bacteria among aquifers acting as a travelling seed bank (Griebler et al., 2014). For these monitoring approaches, water grab samples were previously found to give access to snapshots of the diversity found within an aquifer (Voisin et al., 2018), whereas aquifer biofilms developing on artificial surfaces (clay beads) were shown to be more integrative and informative of the groundwater microbiological quality (Mermillod-Blondin et al., 2019). Clay bead biofilms were found to capture the most abundant aquifer taxa and taxa that could not be detected from grab samples. However, some bacterial taxa were still not detectable by this approach because of a poor ability at colonizing clay beads over short time periods. A field-based investigation was thus performed to further explore the relative contributions of a set of sources such as runoffs and urban soils on the observed biofilm assemblages recovered from an aquifer. A Bayesian methodology, named SourceTracker (Knights et al., 2011), was used to investigate community coalescence from 16S rRNA gene-based DNA metabarcoding datasets. Complementary datasets were then assembled from an additional DNA marker named tpm (encoding EC:2.1.1.67, which catalyzes the methylation of thiopurine drugs) (Favre-Bonté et al., 2005). This genetic marker enables finer taxonomic allocations down to the species level to explore the coalescence of a set of bacterial species and subspecies, including plant and human pathogens, within the aquifer microbial community.
2.1 Experimental site
The Chassieu urban catchment is located in the suburbs of Lyon (France). It has a surface of 185 ha and hosts mainly industrial and commercial activities (i.e., wholesaling, recovery and waste management, metal surface treatment, car wash, and repair services). The imperviousness coefficient of the catchment area is approximately 75 %. No significant modifications impacting the urban watershed were recorded during the investigation. Stormwater and dry-weather flows from industrial activities are drained by a network separated from the sewer. This network transfers waters into the Django-R (Django Reinhardt) SIS, which is a part of the OTHU (Observatoire de Terrain en Hydrologie Urbaine) long-term experimental observatory dedicated to urban waters (http://www.graie.org/othu/, last access: May 2020). This SIS contains an open and dry detention basin (DB) (32 000 m3) built on a concrete slab, with edges impermeabilized by a thick plastic lining. This DB allows for a settling of coarse- and medium-size particles, resulting in sedimentary deposits which favour plant cover development. The DB water content is delivered within 24 h into an infiltration basin (IB) (61 000 m3), which favours the recharge of the connected aquifer (AQ). This infiltration basin had a vadose zone of about 11 m during the investigation, and its geology, hydrology, ecology and pollution levels had been previously investigated (e.g., Barraud et al., 2002; Le Coustumer and Barraud, 2007; El-Mufleh et al., 2014).
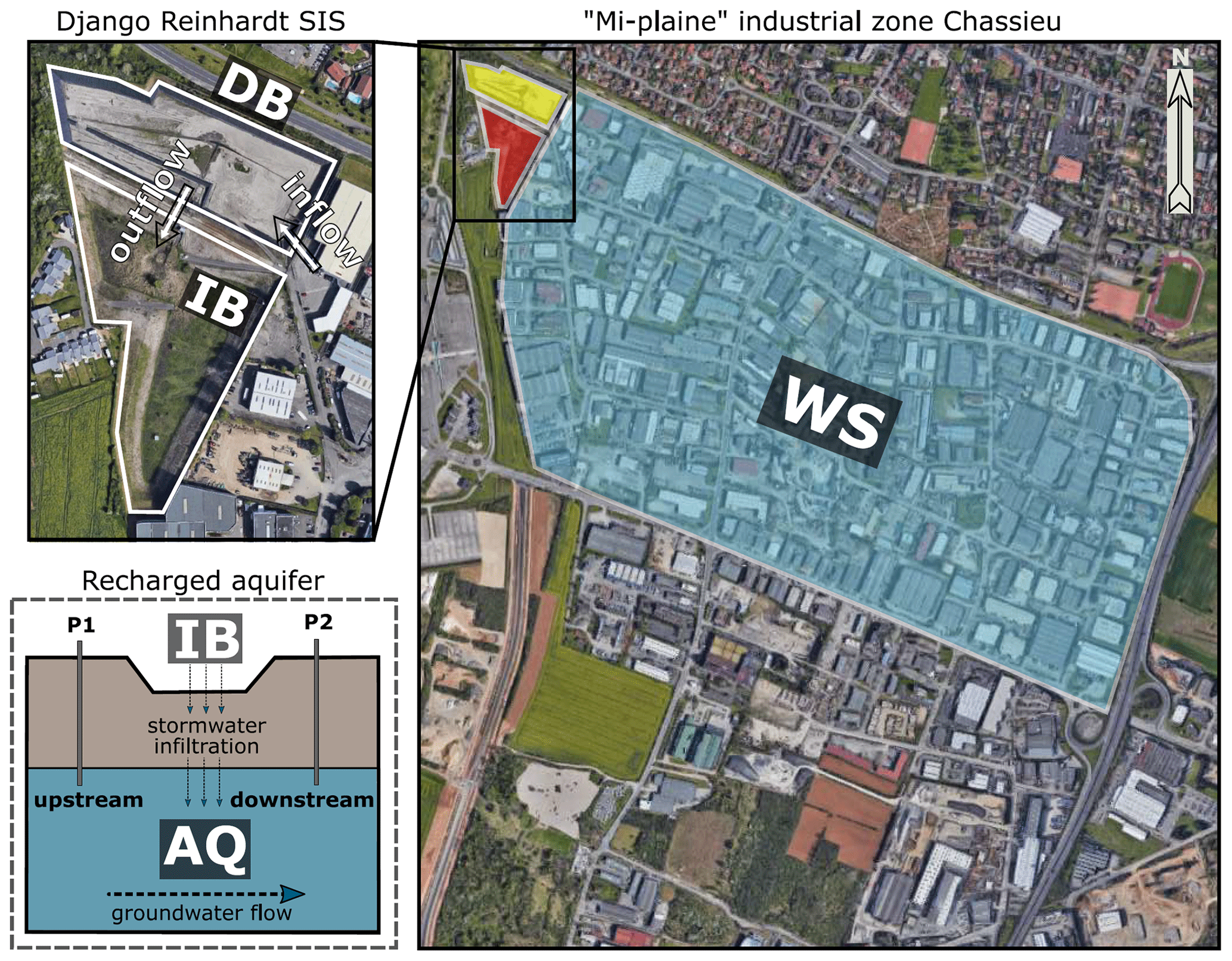
Figure 1Scheme illustrating the trajectory of urban runoffs from the industrial watershed (WS) towards the stormwater infiltration system (SIS) investigated in this study. The urban watershed is located in Chassieu (France). The SIS is made of a detention basin (DB) and an infiltration basin (IB) and is connected to the Lyon 200 km2 east aquifer (AQ). (Map data: © Google and Maxar Technologies.)
The Chassieu watershed, the Django-R SIS and the Lyon aquifer were investigated in this study (Fig. 1 and Table S1). Watershed (WS) runoff waters have been collected from sampling points spread over the catchment (21 subwatersheds over three sampling periods, n=64 samples). Sediments from the detention basin (hereafter DB) were recovered from a 50 cm2 area covering the full sediment column down to the concrete slab (n=20 samples). These sediments (or urban soils) often had an herbaceous plant cover and were sampled in four areas defined according to the hydrological forces prevailing in the basin (e.g., Marti et al., 2017; Sébastian et al., 2014). Infiltration basin (hereafter IB) soil samples were collected from three zones (the area receiving the inflow waters, the bottom area of the basin and an upper zone of the basin exposed to inflow waters only during heavy-rain events) (n=5 samples per zone) at a 0–10 cm depth covering a surface of 50 cm2. The aquifer samples were recovered from piezometers located upstream (up; in a zone of the aquifer not influenced by water recharge) and downstream (dw; in a zone of the aquifer influenced by water recharge) of the SIS of the Django-R site at a depth of 2 m below the water table (e.g., Barraud et al., 2002; Voisin et al., 2018) (Fig. 1). Groundwater samplings (n=6; named AQ_wat) were performed with an immerged pump, used at a pumping rate of 6–8 L min−1 (PP36 inox, SDEC, Reignac-sur-Indre, France) and previously cleaned with 70 % ethanol. The first 50 L were used to rinse the sampling equipment and were subsequently discarded. The following 6 L was used for the microbiological analyses. The biofilm samples (AQ_bio) from the aquifer were recovered from clay beads incubated in the aquifer over 10 d using the piezometers described above (n=6 samples). Clay beads were used as physical matrices to sample groundwater biofilms according to Voisin et al. (2016).
2.2 DNA extractions, 16S rRNA gene qPCR (quantitative polymerase chain reaction) assays and PCR product DNA sequencing
Approximately 600 mg of sediments or soils or up to 5 L of aquifer or runoff water samples filtered using 0.22 µm polycarbonate filters was used per DNA extraction. Total DNA was extracted from soils and sediments or filters using the FastDNA SPIN® Kit for Soil (MP Biomedicals, Illkirch, France). For clay bead biofilms, microbial cells were detached by shaking at 2500 rpm for 2 min in 10 mL of 0.8 % NaCl. These suspensions were then filtered, and their DNA contents were extracted as indicated above. Blank samples were performed during these extractions for both the soils and sediments or filtered cells. DNA was quantified using a NanoDrop UV-Vis (ultraviolet–visible) spectrophotometer. Blank DNA extracts showed values below the detection limit. DNA extracts were visualized after electrophoresis at 6 V cm−1 using a TBE buffer (89 mM tris, 89 mM boric acid and 2 mM ethylenediaminetetraacetic acid – EDTA; pH 8.0) through a 0.8 % (w∕v; weight by volume) agarose gel and DNA staining with 0.4 mg mL−1 ethidium bromide. A Gel Doc XR+ System (Bio-Rad, France) was used to observe the stained DNA and confirm their relative quantities (between 20 and 120 ng µL−1; median value around 40 ng µL−1) and qualities. DNA was kept at −80 ∘C and shipped on ice within 24 h to the DNA sequencing services when appropriate.
Quantitative PCR assays were performed on the DNA extracts to estimate their relative content in 16S rRNA gene copies. These assays were performed on a Bio-Rad CFX96 real-time (RT) PCR instrument with Bio-Rad CFX Manager software, version 3.0 (Marnes-la-Coquette, France). The 16S rRNA gene primers 338F and 518R described by Park and Crowley (2006) were used, together with the Brilliant II qRT-PCR SYBR Green Low ROX Master Mix for SYBR Green qRT-PCR. The melting temperature was 60 ∘C. Linearized plasmid DNA containing a 16S rRNA gene was used as a standard and obtained from Marti et al. (2017). Presence of inhibitors in the DNA extracts was checked by spiking a known amount of plasmids harbouring int2 (107 copies of plasmid per microlitre) in the PCR mix. A number of cycles needed to get a PCR signal was compared with wells where only plasmid DNA harbouring int2 was added to the qPCR mix. When a high number of cycles was needed to observe a signal, a 5- or 10-fold dilution of the DNA extract was done, and another round of tests was performed to confirm the absence of PCR inhibitions. Each assay was triplicated on distinct DNA extracts, and technical triplicates were performed. The 16S rRNA gene qPCR datasets are presented in Fig. S1 in the Supplement. These assays confirmed the high number of bacterial cells per compartment (Fig. S1 and Table S2): (1) soils from the infiltration basin (IB) had a median content of 1.32×1011 16S rRNA gene copies per gram dry weight; (2) sediments from the detention basin (DB) had 1.83×1011 16S rRNA gene copies per gram dry weight; (3) the runoff waters (WS) had a median content of 4.75×108 16S rRNA gene copies per millilitre; (4) the aquifer waters (AQ_wat) had 3.10×106 16S rRNA gene copies per millilitre; and (5) the aquifer clay bead biofilms showed 1.35×107 16S rRNA gene copies per square centimetre.
Sequencing of V5-V6 16S rRNA gene (rrs) PCR products were performed by MR DNA sequencing services (Shallowater, Texas, USA) on an Illumina MiSeq v3. The PCR products were generated using DNA primers 799F (barcode + ACCMGGATTAGATACCCKG) and 1193R (CRTCCMCACCTTCCTC) reported by Beckers et al. (2016). PCR amplifications were performed using the HotStarTaq Plus Master Mix Kit (QIAGEN, USA) using the following temperature cycles: 94 ∘C for 3 min, followed by 28 cycles of 94 ∘C for 30 s, 53 ∘C for 40 s and 72 ∘C for 1 min, with a final elongation step at 72 ∘C for 5 min. PCR products and blank control samples were verified using a 2 % agarose gel and following the electrophoretic procedure described above. PCR products obtained from field samples showed sizes around 430 bp (base pair), but blanks did not show detectable and quantifiable PCR products. Dual-index adapters were ligated to the PCR fragments using the TruSeq® DNA Library Prep Kit which also involved quality controls of the ligation step (Illumina, Paris, France). Illumina MiSeq DNA sequencings of the PCR products were paired-end and set up to obtain around 40 000 reads per sample.
The tpm DNA libraries were also sequenced by the Illumina MiSeq v3 technology but by the Biofidal DNA sequencing services (Vaulx-en-Velin, France). PCR products were generated using the following mix of degenerated PCR primers: ILMN-PTCF2 (5'–P5 adapter tag + universal primer + GTGCCGYTRTGYGGCAAGA-`3), ILMN-PTCF2m (5'–P5 adapter tag + universal primer + GTGCCCYTRTGYGGCAAGT-`3), ILMN-PTCR2 (5'–P7 adapter tag + universal primer + ATCAKYGCGGCGCGGTCRTA-`3) and ILMN-PTCR2m (5'–P7 adapter tag + universal primer + ATGAGBGCTGCCCTGTCRTA-`3) targeting conserved regions defined by Favre-Bonté et al. (2005). The universal primer was 5'-AGATGTGTATAAGAGACAG-`3. The P5 adapter tag was 5'-TCGTCGGCAGCGTC-`3. The P7 adapter tag was 5'- GTCTCGTGGGCTCGG-`3. PCR reactions were performed using the 5X Hot BIOAmp® master mix (Biofidal, France) containing 12.5 mM MgCl2 and 10 % DMSO – dimethyl sulfoxide – and 50 ng sample DNA final concentrations. PCR cycles were as follows: (1) a hot start at 94 ∘C for 5 min, (2) 35 cycles consisting of 94 ∘C for 30 s, 58 ∘C for 30 s and 72 ∘C for 30 s, and (3) a final extension of 5 min at 72 ∘C. The mix had two carefully optimized enzymes, the HOT FIREPol® DNA polymerase and a proofreading polymerase. This enzyme blend has both 5' → 3' exonuclease and 3' → 5' proofreading activities. This mix exhibits an increased fidelity (up to 5-fold) compared to a regular Taq polymerase. PCR products and blank control samples were verified using a 2 % agarose gel and following the electrophoretic procedure described above. PCR products obtained from field samples showed sizes around 320 bp, but blanks did not show detectable and quantifiable PCR products. Index and Illumina P5 or P7 DNA sequences were added by Biofidal through a PCR procedure using the same Hot BIOAmp® master mix and the above temperatures but limited to 15 PCR cycles. Indexed P5- and P7-tagged PCR products were purified using the SPRIselect procedure (Beckman Coulter, Roissy, France). PCR products and blank control samples were verified using the QIAxcel DNA kit (QIAGEN, France), and band sizes around 400 bp were observed but not in the blank samples. Quantification of PCR products by the PicoGreen approach using the QuantiFluor dsDNA (double-stranded) kit (Promega, France) and a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, France) was performed and showed low values among the blanks, which were at the limit of detection (around 0.07 ng µL−1). Still, with tpm-harbouring bacteria being in low number among a bacterial community (about 2 %–3 %), these controls were run during the MiSeq DNA sequencing of the PCR products. Illumina MiSeq DNA sequencings of the tpm PCR products were pair-ended and set up to obtain around 40 000 reads per sample. Blank samples generated low numbers of tpm reads (blank 1=24 reads; blank 2=3 reads, blank 4=1028 reads and blank 5=1 read), and these have been listed in Table S3. These reads mainly belonged to unknown species (86 %). However, reads from Pseudomonas fluorescens (from OTUs – operational taxonomic units – not found in the field samples), P. xanthomarina (17 reads over all blanks) and P. fragi (n=3 reads over all blanks) were recovered but did not have any impact on the coalescence analysis. The 16S rRNA and tpm gene sequences reported in this work are available at the European Nucleotide Archive (https://www.ebi.ac.uk/ena, last access: May 2020).
2.3 Bioinformatic analyses
All paired-end MiSeq reads were processed using Mothur 1.40.4 by following a standard operation protocol (SOP) for MiSeq-based microbial community analysis (Schloss et al., 2009; Kozich et al., 2013), the so-called MiSeq SOP available at http://www.mothur.org/wiki/MiSeq_SOP (last access: May 2020). Due to the large number of sequences to be processed, the “cluster.split” command was used to assign sequences to OTUs. For the 16S rRNA (rrs) gene sequences, reads were filtered for length (>300 bp), quality score (mean ≥25), number of ambiguous bases (=0) and length of homopolymer runs (<8) using the “trim.seqs” script in Mothur, and singletons were discarded. The 16S rRNA gene sequences passing these quality criteria were aligned to the SILVA reference alignment template (release 128; https://www.arb-silva.de, last access: May 2020). Unaligned sequences were removed. Chimeric sequences were identified using the “chimera.uchime” command and removed. Variability in the number of cleaned reads per sample was observed but not correlated with variations in the number of 16S rRNA gene sequences (Table S2). These variations were thus considered to be due to the DNA sequencing process. Therefore, a subsampled dataset (20 624 reads per sample, with the exclusion of samples with total reads below this threshold) was used to mitigate the artifact of sample library sizes. Operational taxonomic units (OTUs) were defined using a 97 % identity cutoff as recommended by several authors in order to collapse sequences into groups that reduce the incidence of sequence errors on the datasets (e.g., Eren et al., 2013; Johnson et al., 2019). It is to be noted that amplicon sequence variants (ASVs) could also be used to build contingency tables (e.g., Callahan et al., 2016; Karstens et al., 2019). However, exact sequence variants can generate uncertainties when using 16S rRNA gene sequences because of variations among species and strains due to the presence of multiple copies per genome (Johnson et al., 2019). Figure S2 shows the OTU rarefaction curves for the full and the subsampled datasets. The subsampled dataset was used for all downstream analyses except those of the SourceTracker Bayesian approach (see below). OTUs were affiliated to taxonomic groups by comparison with the SILVA reference alignment template if a bootstrap p value over 80 % was obtained. FAPROTAX (Functional Annotation of Prokaryotic Taxa; Louca et al., 2016) functional inferences were performed on the MACADAM (MetAboliC pAthway DAtabase for complex Microbial community function analysis) Explore website (http://macadam.toulouse.inra.fr/, last access: May 2020) according to Le Boulch et al. (2019). For the tpm gene sequences, chimeric sequences and primers, barcodes were removed, and the dataset was limited to sequences of a minimum length of 210 bp (average length = 215 bp). These reads were aligned against the tpm database (BD_TPM_Mar18_v1.unique_770seq). Unaligned sequences were removed. The number of sequences per sample was then subsampled (4636 sequences per sample, with the exclusion of samples with total reads below this threshold). Operational taxonomic units (OTUs) were defined at a 100 % identity cutoff. The BD_TPM_ Mar18_v1.unique_770seq database (http://www.graie.org/othu/donnees, last access: May 2020) was used to classify the sequences using the Wang text-based Bayesian classifier (Wang et al., 2007) and a p bootstrap value above 80 %. Local BLAST analyses were performed on the BD_TPM_Mar18_v1.unique_770seq database using the NCBI (National Center for Biotechnology Information) BLAST (Basic Local Alignment Search Tool, version X) program to check the quality of the taxonomic affiliations.
2.4 Statistical analyses
All statistical analyses were performed in R (v3.5.1). For the 16S rRNA gene sequences, alpha-diversity estimates were computed using the function “rarefy” from the “vegan” package (Oksanen et al., 2015). Richness (Sobs) was computed as the number of observed OTUs in each sample. The diversity within each individual sample was estimated using the nonparametric Shannon index. To estimate whether the origin of the samples influenced the alpha diversity, an ANOVA test with Tukey post hoc tests was performed. Shared and unique OTUs were depicted in Venn diagrams with the “limma” package (Ritchie et al., 2015). Concerning the beta-diversity analyses, a neighbour-joining tree was constructed with a maximum-likelihood approximation method using FastTree (Price et al., 2009). Weighted UniFrac distances were calculated for all pairwised OTU patterns according to Lozupone et al. (2011) and used in a principal coordinates analysis (PCoA) (Anderson and Willis, 2003). Permutation tests of distances (PERMANOVAs) (Anderson, 2001) were performed using the vegan package (Oksanen et al., 2015) to establish the significance of the observed groupings.
2.5 Analyses of bacterial community coalescence
The SourceTracker computer package (Knights et al., 2011) was used to investigate community coalescence. SourceTracker is a Bayesian approach built to estimate the most probable proportion of user-defined “source” DNA reads in a given “sink” community. In the present analysis, various scenarios of community coalescence were investigated such as the coalescence of bacterial taxa from the watershed runoff waters and sediments from the detention and infiltration basins with those of the downstream-SIS aquifer water samples or of recent biofilms developing on clay beads incubated in the aquifer. SourceTracker was run with the default parameters (rarefaction depth =1000 reads from the original cleaned dataset of 16S rRNA gene reads; Fig. S2a; burn-in: 100, restart: 10) to identify sources explaining the OTU patterns observed among the aquifer samples (waters and clay bead biofilms, n=12). Alpha values were tuned using cross-validation (alpha 1=0.001 and alpha 2=1). The relative standard deviation (RSD) based on three runs was used as a gauge to evaluate confidence on the computed values (Henry et al., 2016; McCarthy et al., 2017).
3.1 16S rRNA V5-V6 gene sequence distribution biases and profiling
The analysis of the 16S rRNA V5-V6 gene libraries yielded 2 124 272 high-quality sequences distributed across 103 samples as described in Table S2. Subsampling-based normalization was applied (20 624 reads per sample), and sequences were distributed into 10 231 16S rRNA gene OTUs (with >97 % identity between sequences of an OTU). The rarefaction curves are shown in Fig. S2. At all sampling sites, bacterial communities were dominated by Proteobacteria, Bacteroidetes and Actinobacteria (WS =95 % of total reads, DB =84 %, IB =71 %, AQ_bio =99 %; AQ_wat =59 %), but 10 other phyla with relative abundances greater than 0.5 % were also detected (Fig. 2a and Table S4). Alpha-diversity estimates showed that aquifer samples harboured a microbiome with a significantly lower richness (AQ_bio: Sobs=278 OTUs ±106; AQ_wat: Sobs=490 OTUs ±333) and a less diverse bacterial community (AQ_bio: H' and AQ_wat: H' ) than the ones of the upper compartments ( OTUs ±232; OTUs ±245, OTUs ±177 and H'; H' and H') (ANOVA, p<0.001) (Fig. 2b and Table S5). Among the surface samples, a greater diversity was observed among the soil samples from the infiltration basin than from samples of watershed runoff waters and sediments recovered from the detention basin (ANOVA, p<0.05). In the aquifer, water grab samples were more diverse and showed higher 16S rRNA gene OTU contents than biofilms recovered from the clay beads incubated over a 10 d period (ANOVA, p<0.05) (Fig. 2b and Table S5).
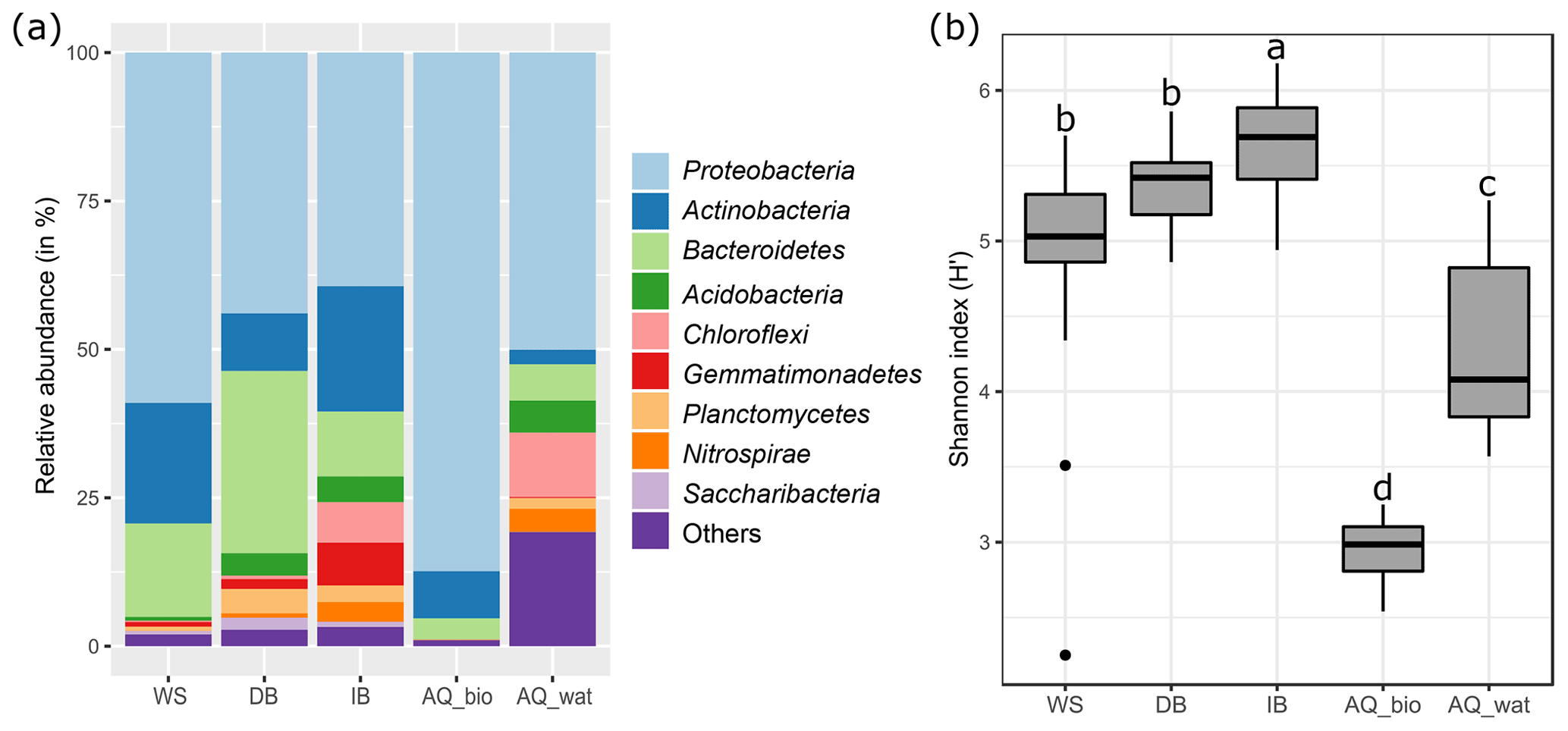
Figure 2General features of the V5-V6 16S rRNA gene metabarcoding DNA sequences obtained from runoff, SIS and aquifer samples. See Fig. 1 for a description of the experimental design. Panel (a) illustrates the relative abundance of the main bacterial phyla observed per compartment, and panel (b) shows boxplots illustrating the variations observed per compartment between the Shannon diversity indices computed from the V5-V6 16S rRNA gene OTU contingency table (subsampled dataset). A one-way ANOVA test with multiple Tukey post hoc tests was performed to investigate the differences between compartments. Different letter codes indicate significant differences (p<0.05). WS: runoff waters from the watershed; DB: sediments from the detention basin; IB: soils from the infiltration basin; AQ_wat: aquifer waters; AQ_bio: aquifer clay beads biofilms.
The structure of bacterial communities inferred from V5-V6 16S rRNA gene sequences changed markedly along the watershed. A PCoA ordination of the OTU profiles based on weighted UniFrac distances showed samples to be clustered according to their compartment of origin (i.e., WS, DB, IB, AQ_bio and AQ_wat) (Fig. 3). These changes in community structures between compartments were supported by PERMANOVA statistical tests (F=20.7, p<0.001). Bacterial communities per compartment were found to contain core and flexible (defined as not conserved between all sampling periods) bacterial taxa. Within the same compartment, similarities between bacterial community profiles ranged from 65 % (AQ_wat) to 82 % (IB), whereas similarities across compartments ranged from 48 % (DB vs. AQ_bio) to 66 % (DB vs. IB) (Fig. S3). Bacterial community profiles of the aquifer waters were found closer to the ones of the detention basin deposits (57 %) and soils of the infiltration basin (61 %) than those of the aquifer biofilms (48 % and 49 %, respectively). However, more than 89 % of the 16S rRNA gene OTUs (n=8284) identified above the aquifer (WS, DB and IB) were not detected in groundwater samples (AQ_bio and AQ_wat) (Fig. S4). This large group of OTUs was made of minor taxa which accounted for 37 %, 44 % and 47 % of the total reads recovered from the WS, DB and IB samples, respectively.
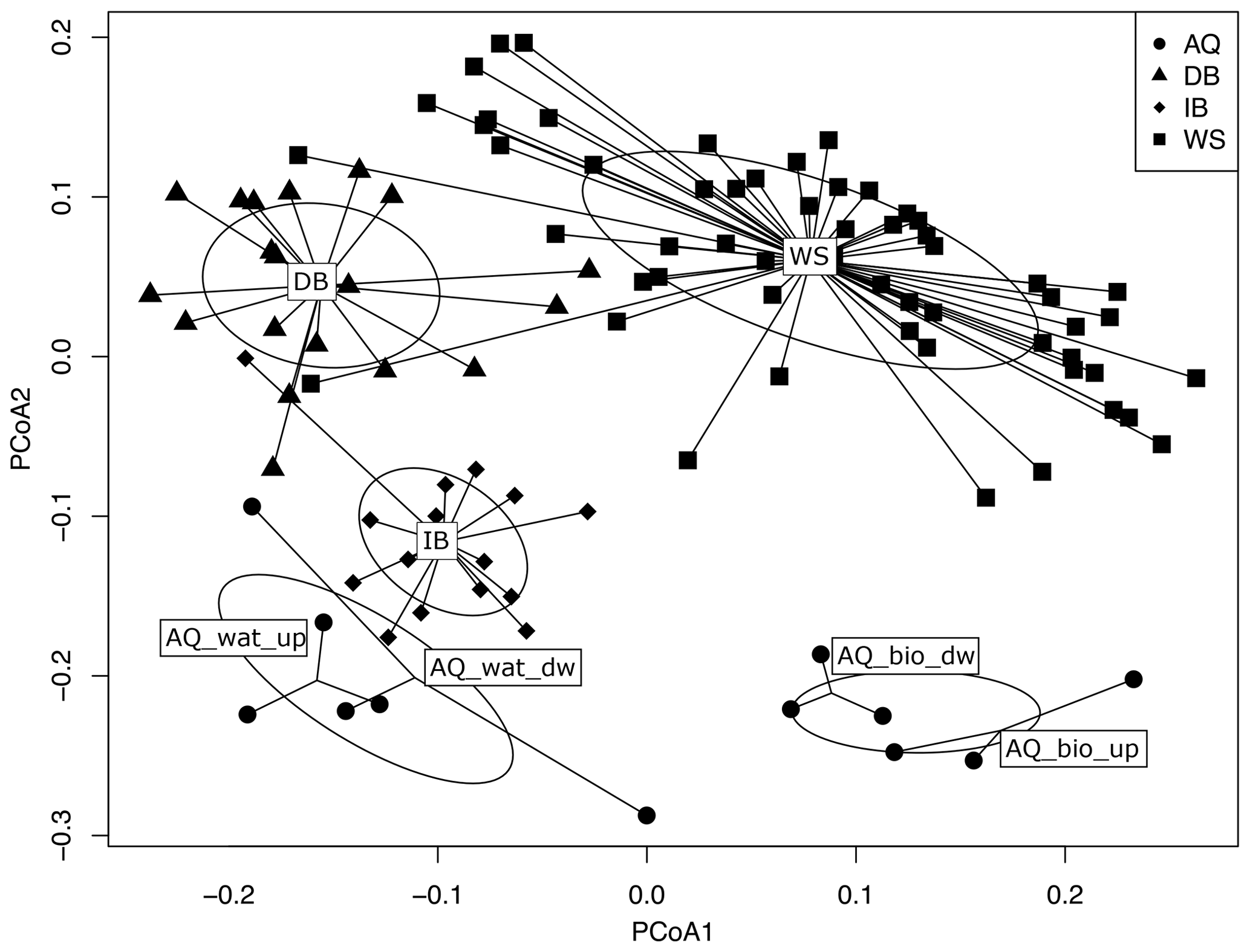
Figure 3PCoA analysis of weighted UniFrac dissimilarities between the V5-V6 16S rRNA gene OTU profiles of the watershed (WS) runoff waters, urban sediments and soils from the connected detention (DB) and infiltration (IB) basins, and waters (AQ_wat) and biofilms (AQ_bio) from the connected aquifer. See Fig. 1 for a description of the experimental design. Ellipses are representative of the variance observed (standard error) between the ordinations of a group of samples. PERMANOVA tests confirmed the significance (p<0.001) of the groupings. The proportion of the eigenvalue per axis over the sum of all eigenvalues was 25.0 % for PCoA1 and 17.6 % for PCoA2. The subsampled V5-V6 16S rRNA gene dataset was used for the computations.
3.2 Coalescence of surface and aquifer bacterial communities
3.2.1 Source-tracking analyses from the 16S rRNA gene sequences
A SourceTracker analysis was performed to estimate the coalescence of bacterial taxa inferred from V5-V6 16S rRNA gene reads from the watershed and SIS down into the aquifer waters and biofilm bacterial communities. This analysis indicated significant coalescence between the bacterial communities of the runoffs, the soils and sediments of the SIS, and the aquifer samples. The aquifer water microbial community upstream of the SIS was found to explain about 40 % of the downstream water microbial community (Table 1), while 16S rRNA gene reads from the runoff waters were found to explain about 5 %, and those of the DB were found to be around 8 % of the observed patterns (Table 1). The infiltration basin explained about 7 % of the observed diversity among the SIS-impacted aquifer water community. The aquifer biofilm bacterial communities were also found to be assemblages of communities from the surface environments. The origin of more than 94 % of the SIS-impacted aquifer biofilms could be explained by the SourceTracker. Main sources of taxa were inferred to be the upstream aquifer waters (59 %), the sediments of the detention basin (22 %) and the runoff waters (8.5 %) (Table 1). Soils from the infiltration basin did not appear to have contributed substantially to the taxa recovered from these aquifer biofilms (<4 %) (Table 1). Aquifer biofilms recovered upstream of the SIS showed a high proportion of taxa related to those observed among the runoff waters (44 %) and the aquifer waters (49 %). This was not considered surprising because runoff infiltration can occur in several sites upstream of the SIS (although no direct relation with other SISs could be made so far).
Table 1Coalescence of surface and aquifer bacterial communities inferred by the SourceTracker Bayesian approach and the 16S rRNA gene metabarcoding dataset*.
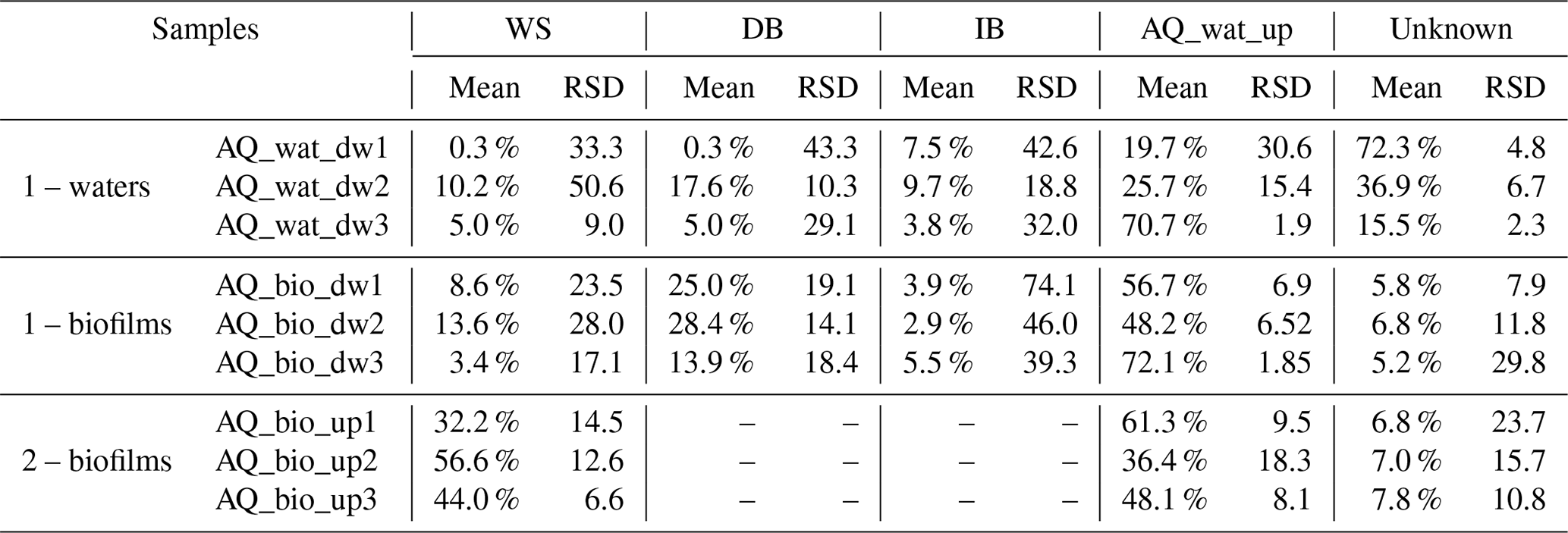
* Two main groups of analyses are shown from subsampled datasets set at 1000 reads: (1) reads from WS, DB, IB and aquifer waters from upstream of the SIS were considered as the sources of taxa for the aquifer samples downstream of the SIS; (2) reads from WS and the aquifer waters upstream of the SIS were considered as the sources of taxa for the aquifer biofilms recovered upstream of the SIS. SourceTracker was run three times using the 16S rRNA gene OTU contingency table and the default parameters. Relative contributions of the sources were averaged. Relative standard deviations (%RSD) are indicated and used as confidence values. RSD >100 % indicates low confidence of the estimated value. WS: watershed runoff waters; DB: detention basin sediments; IB: infiltration basin sediments; AQ_wat: aquifer waters. Sequences that could not be attributed to one of the tested sources were grouped under the term unknown.
3.2.2 16S rRNA gene-inferred bacterial taxa undergoing coalescence in the aquifer
To identify the bacterial taxa involved in the coalescence process, OTUs of the 16S rRNA gene dataset were allocated to taxonomic groups using the SILVA reference alignment template. These taxonomic allocations indicated that (1) 14 genera were only recorded in the aquifer samples, (2) 421 genera were only recorded in the upper-surface compartments of the watershed and (3) 219 were recorded among aboveground and aquifer compartments (Table S6). The following bacterial genera were exclusively associated with the aquifer bacterial communities: Turicella, Fritschea, Metachlamydia, Macrococcus, Anaerococcus, Finegoldia, Abiotrophia, Dialister, Leptospirillum, Omnitrophus, Campylobacter, Sulfurimonas, Haemophilus and Nitratireductor. These bacterial genera were recovered from all water samples, and five were also detected in biofilms (Table S6). These genera were associated with 926 16S rRNA gene OTUs that accounted for, respectively, 48 % and 1.8 % of the total reads recovered from aquifer waters and aquifer biofilms developing on clay beads. FAPROTAX functional inferences indicated some of these genera to be host-associated such as Fritschea, Metachlamydia, Finegoldia, Campylobacter and Haemophilus, with the latter two being well known to contain potential pathogens. Campylobacter and Sulfurimonas cells have also been associated with nitrogen and sulfur respiration processes, and Leptospirillum has been associated with nitrification.
Regarding the bacterial taxa of the aboveground communities matching those of the aquifer samples, a total of 1021 16S rRNA gene OTUs was found to be shared between these compartments (Table 2 and Fig. S4). These OTUs consisted of abundant taxa, as they accounted for 9.7 %–39.4 % of the total reads for the samples recovered from the surface compartments and for 33.6 %–83.4 % and 95.0 %–99.4 % of the total reads of the water and biofilm aquifer samples, respectively (Table 2). The Beta- and Gammaproteobacteria dominated this group. It is noteworthy that aquifer samples collected upstream of the SIS shared fewer OTUs with the surface compartments (125 OTUs ±41) than samples under the influence of the infiltration system (332 OTUs ±85) (Table 2 and Fig. S4). The shared OTUs between aquifer samples and the upper compartments represented a higher fraction of bacterial communities in samples recovered downstream of the SIS (81.3 %±22.8 of total reads) compared to those collected upstream (68.9 %±30.9 of total reads) (Table 2). Reads from Pseudomonas, Nitrospira, Neisseria, Streptococcus and Flavobacterium were the most abundant (>1 %) of the shared OTUs recovered in the aquifer water samples, whereas those allocated to Pseudomonas, Duganella, Massilia, Nocardia, Flavobacterium, Aquabacterium, Novosphingobium, Sphingobium, Perlucidibaca and Meganema were the most abundant (>1 %) among the aquifer biofilms (Table S6). Most of these aquifer water taxa (except Streptococcus) were found to be involved in denitrification or nitrification as inferred from FAPROTAX. The biofilm taxa were most often associated with hydrocarbon degradation (Novosphingobium, Sphingobium and Nocardia) by FAPROTAX. Several of these biofilm bacterial genera were also found to be containing potential human pathogens (Duganella, Massilia, Nocardia and Aquabacterium) by FAPROTAX (and published clinical records). A set of 14 potentially hazardous bacterial genera was selected from Table S6 and used to illustrate the coalescence of bacterial taxa among the aquifer samples in Fig. 4. The 16S rRNA gene reads from Flavobacterium prevailed in all upper compartments (WS =6.9 % of total reads, DB =13.4 % and IB =8.3 %) and were a significant amount in the connected aquifer (AQ_wat =1.1 % and AQ_bio =3.1 %) (Fig. 4b and Table S6). Pseudomonas 16S rRNA gene reads were in relatively lower numbers in the upper compartments (WS =0.4 % of total reads, DB =0.4 % and IB <0.05 %) than the aquifer (AQ_wat =8.4 % and AQ_bio =35.5 %) (Fig. 4b and Table S6). Similar trends were observed for Nocardia and Neisseria OTUs (Fig. 4b). Notably, OTUs exclusively recovered from the upper compartments were mainly allocated to Gemmatimonas (0.2 %–1.6 % of total reads), Geodermatophilus (0.1 %–1.8 %) and Roseomonas (0.1 %–1.0 %) (Table S6).
Table 2Aquifer 16S rRNA gene (rrs) OTUs detected in the upper compartments of the investigated watershed and SIS*.
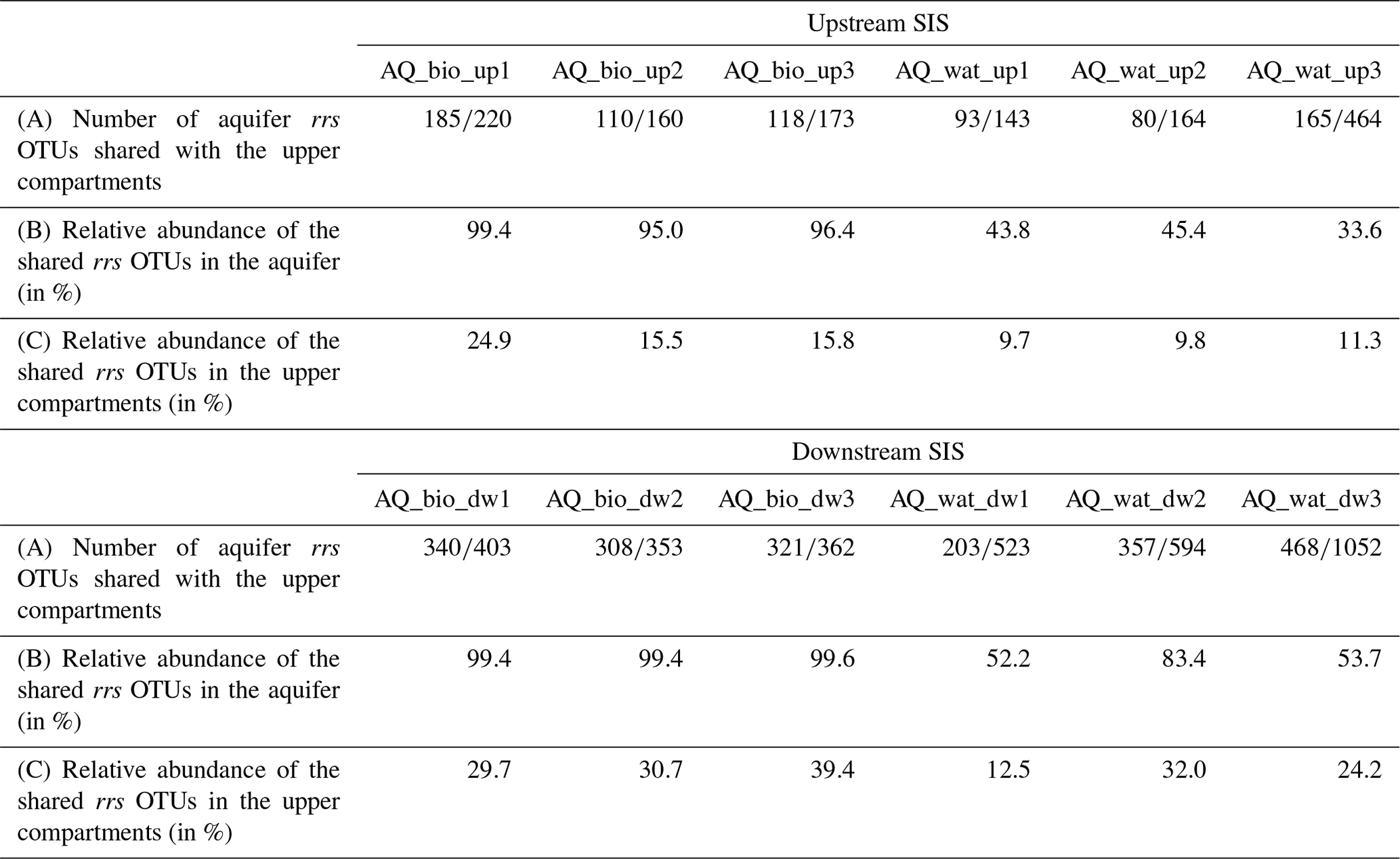
* The number or relative number of shared aquifer rrs OTUs found in the upper compartments (WS, DB and IB) was computed per aquifer sample recovered upstream (up) or downstream (dw) of the SIS (see Fig. 1 for the sampling design), after a resampling of the reads set at 20 624 per sample. AQ_wat: aquifer waters; AQ_bio: aquifer clay beads biofilms; up: upstream of the SIS; dw: downstream of the SIS.
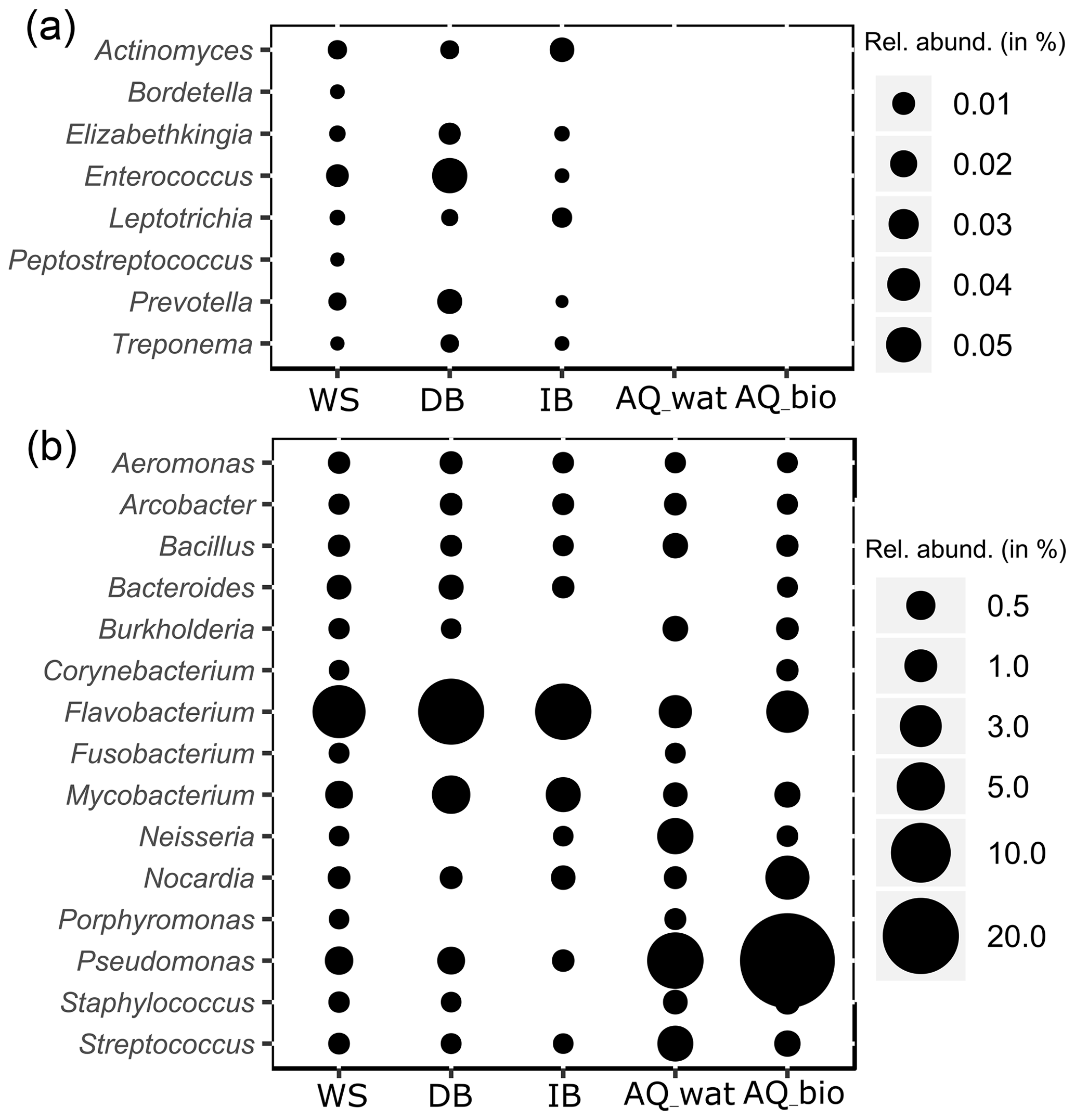
Figure 4Relative numbers of potentially pathogenic bacterial genera along the watershed. The relative abundance (rel. abund.) of bacterial genera exclusively detected in upper compartments (a) or both in the upper compartments and aquifer (b) are presented. Size of bubbles is proportional to the relative abundance (in %) of each bacterial genus per sampled compartment. WS: runoff waters from the watershed; DB: sediments from the detention basin; IB: sediments from the infiltration basin; AQ_wat: aquifer waters; AQ_bio: aquifer clay beads biofilms.
3.2.3 Coalescence of Pseudomonas and other tpm-harbouring bacterial species
DNA sequences from tpm PCR products generated according to Favre-Bonté et al. (2005) allowed for a further exploration of the bacterial species undergoing a coalescence with the aquifer microbiome. A total of 19 129 tpm OTUs were recorded among the samples (from datasets resampled to reach 4636 reads per sample). As expected, these tpm reads were mainly assigned to Proteobacteria (WS =92 % of total reads, DB =87 %, IB =76 %, AQ_wat =83 % and AQ_wat =85 %), but some reads could also be attributed to Bacteroidetes, Nitrospirae and Cyanobacteria (Table S7). These taxonomic allocations allowed for the identification of 24 bacterial genera and 91 species, whose distributions are summarized in Tables S7 and S8. The tpm sequences were mainly allocated to Pseudomonas (WS =36 % of the reads, DB =27 %, IB =7 %, AQ_wat =51 % and AQ_bio =48 %), Aeromonas (WS =1 % of the reads, DB =3 %, IB <0.05 %, AQ_wat =0.07 % and AQ_bio <0.05 %), Xanthomonas (WS =4 % of the reads, DB <0.05 %, IB =1 %, AQ_wat =8 % and AQ_bio <0.05 %), Herbaspirillum (WS =11 % of the reads) and Nitrosomonas (DB =4 % of the reads and IB =0.2 %) (Table S8). Reads related to Pseudomonas were allocated to 50 species, including pollutant-degraders (P. pseudoalcaligenes, P. aeruginosa, P. fragi, P. alcaligenes, P. putida and P. fluorescens), phytopathogens (P. syringae, P. viridiflava, P. stutzeri and P. marginalis) and human-opportunistic pathogens (P. aeruginosa, P. putida, P. stutzeri, P. mendocina and Stenotrophomonas acidaminiphila (Table S9). It is to be noted that blank samples sequenced during the tpm metabarcoding assay revealed 23 Pseudomonas OTUs coming from the DNA extraction kit or generated during the PCR product Illumina sequencing process (Table S3). Only OTU00573 was found to be high in number (867 reads), but this contaminant did not have an impact on the coalescence analysis because of its absence in the underground datasets (Table S10). Other contaminant OTUs did not represent more than 10 times the ones observed in the field samples for identical OTUs, a criterion used to distinguish significant contaminants (Lukasik et al., 2017). In fact, only seven OTUs found among the blanks matched OTUs recovered from the environmental samples, and only two of these could be related to well-defined species, i.e., P. xanthomarina (17 reads among all blanks) and P. fragi (3 reads among all blanks). These reads matched a single OTU over 11 allocated to P. xanthomarina in the environmental samples and 1 OTU over 52 for P. fragi (Table S10). Reads related to Aeromonas were attributed to 11 species, but only those allocated to A. caviae could be recovered from the aquifer and aboveground compartments (Table S9). Reads related to the Xanthomonas were allocated to nine species, but only those allocated to X. axonopodis–X. campestris complex and X. cannabis species were recovered from the aquifer and upper compartments (Table S9). Regarding Pseudomonas, tpm reads allocated to P. jessenii, P. chlororaphis and P. resinovorans were restricted to the aquifer samples. Reads allocated to P. aeruginosa, P. anguilliseptica, P. chengduensis, P. extremaustralis, P. fluorescens, P. fragi, P. gessardii, P. koreensis, P. pseudoalcaligenes, P. putida, P. stutzeri, P. umsongensis and P. viridiflava were recovered from the aquifer and upper compartments (Table S9). FAPROTAX analysis indicated that a significant number of the species detected in the aquifer can not only be involved in denitrification (P. aeruginosa, P. fluorescens, P. putida, P. stutzeri, S. acidaminiphila, X. autotrophicus and P. chlororaphis) or nitrification (Nitrospira defluvii and Nitrosomonas oligotropha) but also in hydrocarbon degradation (P. aeruginosa, P. fluorescens and P. putida). Some of these species were also suggested by FAPROTAX to be human pathogens or invertebrate parasites (e.g., P. chlororaphis and P. aeruginosa).
Table 3Relative distribution of tpm reads per OTU (mean ± SD) shared between the upper compartments and the aquifer that were allocated to well-defined species1.
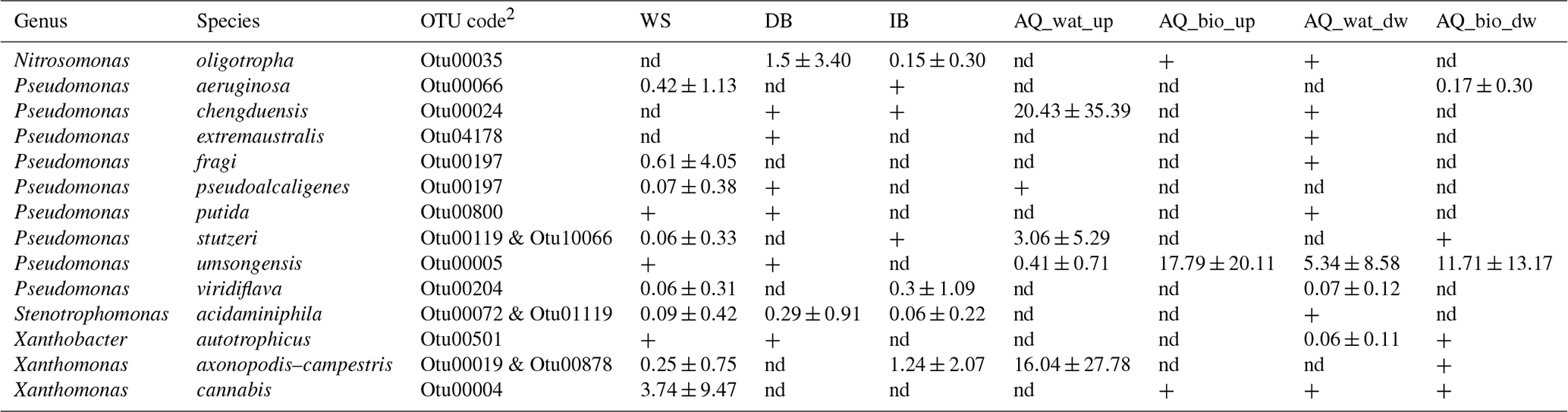
1 All reads from tpm OTUs shared between the upper compartments and the
aquifer were used to compute the relative abundances. 2 tpm sequences of the OTUs are shown in Table S8. WS: watershed runoff
waters; DB: detention basin deposits; IB: soil of the infiltration basin;
AQ_wat: aquifer waters; AQ_bio: aquifer
biofilms. +: OTUs with a relative abundance <0.05 %. nd: not detected.
The tpm OTUs (representative of infra-specific complexes) shared between the upper compartments and the aquifer were allocated to 14 species and 5 genera (Table 3 and Table S10). Four of these OTUs led to higher relative numbers of reads in the aquifer samples, in the following decreasing order: P. umsongensis (Otu00005), P. chengduensis (Otu00024), X. axonopodis–X. campestris (Otu00019 and Otu00878) and P. stutzeri (Otu00119 and Otu10066). These co-occurrences of OTUs between aboveground and aquifer samples support the hypothesis of significant coalescence between these bacterial communities. The other OTUs showed a higher number of reads among the top compartments. The OTU allocated to X. cannabis showed the highest relative number of reads of this group among runoff waters. The distribution pattern of this OTU suggested a relative decline when moving down the aquifer. The P. aeruginosa Otu00066 was recovered in the runoff waters, and biofilms developing on clay beads incubated in the aquifer.
The coalescence of bacterial taxa from runoff and stormwater infiltration systems (SISs) with those of a connected aquifer was investigated. Taxonomic and functional inferences were performed using 16S rRNA gene libraries. In addition, a genetic marker named tpm was used to track species and particular sequence types of Pseudomonas, Aeromonas and Xanthomonas (and a few other genera) from runoffs down into the SIS-impacted aquifer. Estimation of alpha-diversity indices from the 16S rRNA bacterial-community profiling measures indicated that groundwater samples (i.e., waters and biofilms) harboured a less diverse microbiome than those of the top compartments (i.e., WS, DB and IB). A 2- to 5-fold reduction in bacterial richness was observed from the surface compartments down into the aquifer. This result suggested that a high proportion of bacterial taxa carried by stormwater runoffs or thriving in the detention and infiltration basins were retained and/or eliminated by the vadose-zone filtration process. In line with this result, the estimation of the copy number of the bacterial 16S rRNA gene by qPCR revealed that bacterial biomass was much lower in aquifer than in runoff samples. In fact, more than 89 % of the 16S rRNA gene OTUs in the top compartments were not detected in the underground samples. This is in agreement with previous works which have shown that immobilization of microorganisms through porous media is high in the top soil layers and triggered by mechanical straining, sedimentation and adsorption (Kristian Stevik et al., 2004; Krone et al., 1958). Moreover, particles that accumulate as water passes through the soil can form a mat that enhances this straining process (Krone et al., 1958). Despite this filtering effect, infiltration induces significant changes in the diversity of groundwater bacterial communities. Both water and biofilm aquifer samples recovered downstream of the SIS had higher bacterial richness than those collected upstream. It is to be noted that soils of the infiltration basin showed higher bacterial diversity than those of the sediments of the detention basin and runoffs. This is most likely related to a development of plant-associated bacteria in this compartment. Indeed, the infiltration basin was covered by several plant species of Magnoliophyta like Rumex sp. which can disseminate rapidly through rhizomes (Bedell et al., 2013) and generate multiple ecological niches for bacteria.
The SourceTracker Bayesian probabilistic approach based on 16S rRNA gene metabarcoding datasets (Knights et al., 2011) was applied to refine our understanding of the coalescence of microbial communities from aboveground environments down into an aquifer. These inferences revealed variable levels of coalescence in the SIS recharge aquifer depending upon the investigated sink, i.e., waters or biofilms developing on clay beads incubated in the aquifer. Bacterial-community structures of the groundwater samples (upstream and downstream of the SIS) were significantly built from aboveground communities (e.g., those from runoff waters). However, the origin of a high proportion of the diversity observed among the aquifer waters downstream of the SIS remained undefined. This is likely related to the emergence of novel biomes among the vadose zone of a SIS fed with urban waters and pollutants. These biomes would have emerged from the buildup of novel biotopes during the construction and functioning of the SIS (see Winiarski, 2014, for review). The prevailing environmental constraints and pollutants would then have favoured minor taxa (not detectable by DNA metabarcoding approaches) from the aboveground compartments. In fact, chemical pollutants have been shown to be significantly washed off or transported with particles during rain events (El-Mufleh et al., 2014), and some of these were found to reach aquifers fed by SISs (Pinasseau et al., 2019). Among these pollutants, Bernardin-Souibgui et al. (2018) reported that urban sediments found in the detention basin of the experimental site were heavily polluted by polycyclic aromatic hydrocarbons (PAHs). Their contents were estimated at 197±36 ng g dw−1 (dry weight) for light PAHs and at 955±192 ng g dw−1 for heavy PAHs. PCBs (polychlorinated biphenyls) were also recorded for the seven congeners of the European norm for a total of 0.2 to 2.1 mg kg dw−1 (Sebastian et al., 2014). Metallic trace elements (MTEs) were recorded in significant amounts, with Cu being found at about 280 mg kg dw−1, Pb at about 200 mg kg dw−1, Zn at about 1600 mg and Cd at about 5 mg kg dw−1 (Sebastian et al., 2014). MTEs, PCBs and PAHs were also recorded in the soils of the infiltration basin at similar concentrations, e.g., on average, at 0.26 mg PCBs kg dw−1 and at more than 940 ng g dw−1 for PAHs (Winiarski, 2014; Winiarski et al., 2006). These sediments and soils were also found to be contaminated by dioxins at about 36 ng g dw−1 (Winiarski, 2014) and by 4-nonylphenols and bisphenol A, at concentrations varying from 6 to 3400 ng g dw−1 (Wiest et al., 2018). However, MTEs and nonpolar PAHs found among SISs are unlikely to reach groundwaters. To illustrate, Pb and Cd were not recorded at depths below 1.5 m in the non-saturated zone of SISs (Winiarski et al., 2006). In contrast, polar organic pollutants were found to be transferred into aquifers as shown for some pesticides and pharmaceuticals (Pinasseau et al., 2019). These chemical contaminants represent potential energy and carbon sources for microorganisms and can also be detrimental to the growth of some organisms. They can thus have significant impacts on the biology of the contaminated soils and sediments.
Functional inferences from the knowledge on bacterial genera suggested an occurrence of several aquifer taxa involved not only in the nitrogen and sulfur cycles but also in hydrocarbon degradation. Campylobacter, Flavobacterium, Pseudomonas and Sulfurimonas cells have been associated with nitrogen and sulfur respiration processes, and Nitrospira and Leptospirillum have been associated with nitrification. The oligotrophic nature of the aquifer waters (concentrations of biodegradable dissolved organic carbon <0.5 mg L−1; Mermillod-Blondin et al., 2015) is thus likely to have induced a significant selective sorting of microbial taxa among the merged community. Most abundant aboveground taxa often require high energy (organic carbon) and nutrient levels to proliferate (Cho and Kim, 2000; Griebler and Lueders, 2009). Twice as many dissolved organic carbons were detected among aquifer waters of the experimental site recovered downstream of the SIS (1.93 mg L) than upstream (0.88 mg L) (Mermillod-Blondin et al., 2015), and this effect was confirmed for other SISs (Mermillod-Blondin et al., 2015; Winiarski, 2014). Similarly, a large part of the bacterial taxa identified from aquifer biofilms was attributed to aboveground sources by the SourceTracker approach. Indeed, watershed runoff waters and detention basin deposits were found to have significantly contributed to the buildup of the observed biofilm community structures. These biofilms showed a high content of 16S rRNA gene sequences belonging to the Beta- and Gammaproteobacteria. According to the ecological concept of r- and K-selection, these Proteobacteria are often considered as r-strategists, able to respond quickly to environmental fluctuations and colonize more efficiently newly exposed surfaces than other groups of bacteria (Araya et al., 2003; Fierer et al., 2007; Lladó and Baldrian, 2017; Manz et al., 1999; Pohlon et al., 2010). Moreover, because they tend to concentrate nutrients (Flemming et al., 2016), biofilms are likely to favour the survival of such opportunistic bacterial cells capable of exploiting spatially and temporally variable carbon and nutrient sources. Here, taxa recovered from aquifer biofilms were previously recorded to have the ability to use hydrocarbons as carbon and energy sources, e.g., Nocardia, Pseudomonas, Sphingobium and Novosphingobium. As indicated above, SISs and urban runoffs are well known to be highly polluted by such molecules (e.g., Winiarski, 2014; Marti et al., 2017; Wiest et al., 2018). The r- and K-selection ecological concept thus seems to apply to the biofilm community assemblages observed in this work.
Taxonomic allocations of the 16S rRNA OTUs suggested that the aquifer waters and biofilms likely harboured opportunistic human, plant and animal pathogens of the genus Finegoldia, Campylobacter, Haemophilus, Duganella, Massilia, Nocardia, Aquabacterium, Flavobacterium, Pseudomonas, Streptococcus and Aeromonas. A striking observation was the enrichment of 16S rRNA gene reads allocated to Nocardia (about 4 % of the total reads) and Pseudomonas (about 35 % of the total reads) in the biofilms recovered from clay beads incubated downstream of the SIS. Nocardia and Pseudomonas 16S rRNA gene sequences were in much lower relative proportions in the aboveground compartments. The genus Pseudomonas was previously found to be abundant under low-flow conditions and was often associated with biofilm formation (Douterelo et al., 2013). Moreover, Pseudomonas species are well known for their ability to use hydrocarbons as energy and C sources. Regarding the Nocardia cells, there is little known of their ecology, but a few reports indicated a tropism for hydrocarbon polluted urban soils and sediments (e.g., Bernardin-Souibgui et al., 2018; Sébastian et al., 2014). There was no additional approach to further investigate the molecular ecology of Nocardia cells found among the investigated urban watershed. However, a tpm metabarcoding analytical scheme could be applied to DNA extracts to further explore the taxonomic allocations of Pseudomonas and some other tpm-harbouring genera. The applied tpm metabarcoding approach allowed an investigation of the coalescence of about 90 species among the investigated watershed including 50 species of Pseudomonas; 11 species allocated to Aeromonas; and some additional species allocated to Nitrospira, Nitrosomonas, Stenotrophomonas, Xanthobacter and Xanthomonas. A single Aeromonas species, A. caviae, was recorded among the above- and underground environments. More than 10 Pseudomonas species thriving in the recharge aquifer were detected among the aboveground compartments. P. umsongensis and P. chengduensis tpm OTUs were detected aboveground and represented a significant fraction of the tpm-harbouring bacteria retrieved from the aquifer samples. These two species were initially isolated from farm soil and landfill leachates (Kwon et al., 2003; Tao et al., 2014), further supporting the hypothesis that such soil-associated bacteria can migrate through runoff infiltration processes down to natural hydrosystems and can merge with aquifer communities. Regarding the Pseudomonas species that may pose health threats to humans, a tpm OTU affiliated to P. aeruginosa was found to be shared between the surface compartments and the biofilm tpm community developing on clay beads incubated downstream of the SIS. P. aeruginosa thus had the properties allowing for an opportunistic development among the aquifer. This species is known for its metabolic versatility and ability to thrive on hydrocarbons. This is an example of a bacterial r-strategist being able to get established opportunistically in aquifer biofilm communities impacted by urban pollutants. Apart from P. aeruginosa, the species P. putida and P. stutzeri, frequently detected in soils and wastewater treatment plants (e.g., Igbinosa et al., 2012; Luczkiewicz et al., 2015; Miyahara et al., 2010), were also recovered along the watershed and the aquifer. Although these two species were identified in human infections (Fernández et al., 2015; Noble and Overman, 1994), information about their virulence remains scarce. These species are therefore considered to be of less concern than P. aeruginosa and A. caviae, another opportunistic infectious agent (Antonelli et al., 2016) found in the aquifer. P. putida isolates have been shown to be involved in hydrocarbon degradation, and P. stutzeri can play a part in the N cycle either through denitrification or nitrogen fixation.
The knowledge gained from the present study demonstrated that coalescence of microbial communities from an urban watershed with those of an aquifer can occur and yield novel assemblages. Specialized bacterial communities of aquifer waters were slightly reshuffled by the aboveground communities. However, the assemblages observed among recent aquifer biofilms were found to be largely colonized by opportunistic r-strategists coming from aboveground compartments and often associated with the ability to degrade hydrocarbons, e.g., Pseudomonas, Nocardia and Novosphingobium cells. An urban aquifer was found, for the first time, to be specifically colonized not only by species like P. chengduensis, P. umsongensis, P. jessenii, P. chlororaphis and P. resinovorans but also by undesirable human-opportunistic pathogens such as P. aeruginosa and A. caviae. Artificial clay beads incubated in the aquifer through piezometers appeared to be highly efficient trapping systems (termed “germcatchers”) for evaluating the ability of a SIS to prevent the transfer of undesirable r-strategists to an aquifer. Nevertheless, the long-term incidence of allochthonous bacteria on the integrity of aquifer microbiota remains to be investigated.
Free-living aquifer bacterial communities are not likely to be much impaired by exogenous cells. However, microbial communities developing as biofilms on inert surfaces might be significantly reshuffled through selective sorting likely induced, in part, by aboveground chemical pollutants. Microbial biofilms are key structures in the transformation processes of several chemicals and nutrients. They often display much higher cell densities than free-living populations (Crump and Baross, 1996; Crump et al., 1998; van Loosdrecht et al., 1990). Here, we have demonstrated that runoff and SIS bacterial taxa can colonize solid matrices of a deep aquifer. These modified communities could (i) alter geochemical processes which can indirectly impact other groundwater inhabitants, e.g., the amphipod Niphargus rhenorhodanensis and other taxa presented in Foulquier et al. (2011), or (ii) directly impact these inhabitants by inducing a modification of their microbial contents and potentially of their behaviour. The stygofauna feeds on bacteria and is well known to be significantly colonized by bacteria (e.g., Smith et al., 2016). The next step in these studies will be to investigate whether native aquifer biofilm communities can resist repeated invasions by opportunistic r-strategists and if these allochthonous bacteria will impact the ecological health of the stygofauna.
The 16S rRNA gene sequences are available at the European Nucleotide Archive (https://www.ebi.ac.uk/ena, Cournoyer, 2020) using the following accession numbers: PRJEB33510 (IB), PRJEB21348 (DB), PRJEB29925 (AQ) and PRJEB33507 (WS). The tpm gene sequences are available using the accession number PRJEB33622.
The supplement related to this article is available online at: https://doi.org/10.5194/hess-24-4257-2020-supplement.
BC coordinated and designed the experiments. YC, VRN, TW, FMB, RB, LM, RM, FV, EB, DB, JV and BC performed the experiments and contributed to the analysis of the datasets. YC and BC prepared the paper with contributions from all co-authors.
The authors declare that they have no conflict of interest.
We thank the Labex IMU (Intelligence des Mondes Urbains), the Greater Lyon Urban Community, the School of Integrated Watershed Sciences H2O'LYON, the Lyon Urban School and CNRS for their support in the development of interdisciplinary research initiatives such as these. Authors also thank the OTHU network for technical assistance and financial support and the reviewers of this paper for their constructive comments.
This research has been supported by the Agence Nationale de la Recherche (grant nos. ANR-16-CE32-0006, ANR-17-CE04-0010, ANR 17-EURE-0018 and ANR-17-CONV-0004); the Agence Nationale de Sécurité Sanitaire de l'Alimentation, de l'Environnement et du Travail (Iouqmer; grant no. EST 2016/1/120); and the Centre National de la Recherche Scientifique (MITI Urbamic project grant).
This paper was edited by Christian Stamm and reviewed by Louis Carles and one anonymous referee.
Anderson, M. J.: A new method for non-parametric multivariate analysis of variance, Austral. Ecol., 26, 32–46, https://doi.org/10.1111/j.1442-9993.2001.01070.pp.x, 2001.
Anderson, M. J. and Willis, T. J.: Canonical analysis of principal coordinates: a useful method of constrained ordination for ecology, Ecology, 84, 511–525,, 2003.
Antonelli, A., D'Andrea, M. M., Montagnani, C., Bartolesi, A. M., Di Pilato, V., Fiorini, P., Torricelli, F., Galli, L., and Rossolini, G. M.: Newborn bacteraemia caused by an Aeromonas caviae producing the VIM-1 and SHV-12 β-lactamases, encoded by a transferable plasmid, J. Antimicrob. Chemother., 71, 272–274, https://doi.org/10.1093/jac/dkv304, 2016.
Araya, R., Tani, K., Takagi, T., Yamaguchi, N., and Nasu, M.: Bacterial activity and community composition in stream water and biofilm from an urban river determined by fluorescent in situ hybridization and DGGE analysis, FEMS Microbiol. Ecol., 43, 111–119, https://doi.org/10.1111/j.1574-6941.2003.tb01050.x, 2003.
Arnaud, E., Best, A., Parker, B. L., Aravena, R., and Dunfield, K.: Transport of Escherichia coli through a thick vadose zone, J. Environ. Qual., 44, 1424, https://doi.org/10.2134/jeq2015.02.0067, 2015.
Barba, C., Folch, A., Gaju, N., Sanchez-Vila, X., Carrasquilla, M., Grau-Martínez, A., and Martínez-Alonso, M.: Microbial community changes induced by Managed Aquifer Recharge activities: linking hydrogeological and biological processes, Hydrol. Earth Syst. Sci., 23, 139–154, https://doi.org/10.5194/hess-23-139-2019, 2019.
Barnes, K. B., Iii, J. M. M., and Roberge, M. C.: Impervious surfaces and the quality of natural and built environments, Department of Geography and Environmental Planning, Towson University, Baltimore, Maryland, USA, 2001.
Barraud, S., Gibert, J., Winiarski, T., and Bertrand Krajewski, J.-L.: Implementation of a monitoring system to measure impact of stormwater runoff infiltration, Water Sci. Technol., 45, 203–210, 2002.
Beckers, B., Beeck, M., Thijs, S., Truyens, S., Weyens, N., Boerjan, W., and Vangronsveld, J.: Performance of 16S rDNA primer pairs in the study of rhizosphere and endosphere bacterial microbiomes in metabarcoding studies, Front Microbiol., 7, 1–15, https://doi.org/10.3389/fmicb.2016.00650, 2016.
Bedell, J.-P., Mourier, B., Provot, and J., Winiarski, T.: Influences on the establishment and dominance of vegetation in stormwater infiltration basins, Water Sci. Technol., 68, 2576–2583, https://doi.org/10.2166/wst.2013.526, 2013.
Bernardin-Souibgui, C., Barraud, S., Bourgeois, E., Aubin, J.-B., Becouze-Lareure, C., Wiest, L., Marjolet, L., Colinon, C., Lipeme Kouyi, G., Cournoyer, B., and Blaha, D.: Incidence of hydrological, chemical, and physical constraints on bacterial pathogens, Nocardia cells, and fecal indicator bacteria trapped in an urban stormwater detention basin in Chassieu, France, Environ. Sci. Poll. Res., 25, 24860–24881, https://doi.org/10.1007/s11356-018-1994-2, 2018.
Booth, D. B.: Urbanization and the natural drainage system-impacts, solutions, and prognoses, The Northwest Environ. J., 7, 93–118, 1991.
Borchardt, M. A., Bradbury, K. R., Gotkowitz, M. B., Cherry, J. A., and Parker, B. L.: Human enteric viruses in groundwater from a confined bedrock aquifer, Environ. Sci. Technol., 41, 6606–6612, https://doi.org/10.1021/es071110+, 2007.
Callahan, B. J., Sankaran, K., Fukuyama, J. A., McMurdie, P. J., and Holmes, S. P.: Bioconductor workflow for microbiome data analysis: from raw reads to community analyses, F1000Research, 5, 1492, https://doi.org/10.12688/f1000research.8986.2, 2016.
Cho, J.-C. and Kim, S.-J.: Increase in bacterial community diversity in subsurface aquifers receiving livestock wastewater input, Appl. Environ. Microbiol., 66, 956–965, https://doi.org/10.1128/aem.66.3.956-965.2000, 2000.
Chong, M. N., Sidhu, J., Aryal, R., Tang, J., Gernjak, W., Escher, B., and Toze, S.: Urban stormwater harvesting and reuse: a probe into the chemical, toxicology and microbiological contaminants in water quality, Environ. Monit. Assess., 185, 6645–6652, https://doi.org/10.1007/s10661-012-3053-7, 2013.
Crites, R. W.: Micropollutant removal in rapid infiltration, in: Artificial Recharge of Groundwater, Butterworth Publishers, Boston, Massachusetts, 579–608, 1985.
Crump, B. and Baross, J.: Particle-attached bacteria and heterotrophic plankton associated with the Columbia River estuarine turbidity maxima, Mar. Ecol. Prog. Ser., 138, 265–273, 1996.
Crump, B. C., Simenstad, C. A., and Baross, J. A.: Particle-attached bacteria dominate the Columbia River estuary, Aquat. Microb. Ecol., 14, 7–18, 1998.
Cournoyer, B.: 16S rRNA and tpm genes of urban runoffs, and soils and sediments of a stormwater infiltration system, available at: https://www.ebi.ac.uk/ena, last access: 25 August 2020.
Dillon, P., Page, D., Vanderzalm, J., Pavelic, P., Toze, S., Bekele, E., Sidhu, J., Prommer, H., Higginson, S., Regel, R., Rinck-Pfeiffer, S., Purdie, M., Pitman, C., and Wintgens, T.: A critical evaluation of combined engineered and aquifer treatment systems in water recycling, Water Sci. Technol., 57, 753–762, https://doi.org/10.2166/wst.2008.168, 2008.
Douterelo, I., Sharpe, R. L., and Boxall, J. B.: Influence of hydraulic regimes on bacterial community structure and composition in an experimental drinking water distribution system, Water Res., 47, 503–516, https://doi.org/10.1016/j.watres.2012.09.053, 2013.
El-Mufleh, A., Béchet, B., Ruban, V., Legret, M., Clozel, B., Barraud, S., Gonzalez-Merchan, C., Bedell, J.-P., and Delolme C.: Review on physical and chemical characterizations of contaminated sediments from urban stormwater infiltration basins within the framework of the French observatory for urban hydrology (SOERE URBIS), Environ. Sci. Poll. Res., 21, 5329–5346, https://doi.org/10.1007/s11356-013-2490-3, 2014.
Eren, A. M., Vineis, J. H., Morrison, H. G., and Sogin, M. L.: A filtering method to generate high quality short reads using Illumina paired-end technology, PLoS ONE, 8, e66643, https://doi.org/10.1371/journal.pone.0066643, 2013.
Favre-Bonté, S., Ranjard, L., Colinon, C., Prigent-Combaret, C., Nazaret, S., and Cournoyer, B.: Freshwater selenium-methylating bacterial thiopurine methyltransferases: diversity and molecular phylogeny, Environ. Microbiol., 7, 153–164, https://doi.org/10.1111/j.1462-2920.2004.00670.x, 2005.
Fernández, M., Porcel, M., de la Torre, J., Molina-Henares, M. A., Daddaoua, A., Llamas, M. A., Roca, A., Carriel, V., Garzón, I., Ramos, J. L., Alaminos, M., and Duque, E.: Analysis of the pathogenic potential of nosocomial Pseudomonas putida strains, Front. Microbiol., 6, 871, https://doi.org/10.3389/fmicb.2015.00871, 2015.
Fierer, N., Bradford, M. A., and Jackson, R. B.: Toward an ecological classification of soil bacteria, Ecology, 88, 1354–1364, https://doi.org/10.1890/05-1839, 2007.
Flemming, H.-C., Wingender, J., Szewzyk, U., Steinberg, P., Rice, S. A., and Kjelleberg, S.: Biofilms: an emergent form of bacterial life, Nat. Rev. Microbiol., 14, 563–575, https://doi.org/10.1038/nrmicro.2016.94, 2016.
Foulquier, A., Simon, L., Gilbert, F., Fourel, F., Malard, F., and Mermillod-Blondin, F.: Relative influences of DOC flux and subterranean fauna on microbial abundance and activity in aquifer sediments: new insights from 13C-tracer experiments, Freshwater Biol., 55, 1560–1576, https://doi.org/10.1111/j.1365-2427.2010.02385.x, 2010.
Foulquier, A., Malard, F., Mermillod-Blondin, F., Montuelle, B., Dolédec, S., Volat, B., and Gibert, J.: Surface water linkages regulate trophic interactions in a groundwater food web, Ecosystems, 14, 1339-1353, https://doi.org/10.1007/s10021-011-9484-0, 2011.
Griebler, C. and Lueders, T.: Microbial biodiversity in groundwater ecosystems, Freshwater Biol., 54, 649–677, https://doi.org/10.1111/j.1365-2427.2008.02013.x, 2009.
Griebler, C., Malard, F., and Lefébure, T.: Current developments in groundwater ecology – From biodiversity to ecosystem function and services, Current Opinion Biotechnol., 27, 159–167, https://doi.org/10.1016/j.copbio.2014.01.018, 2014.
Henry, R., Schang, C., Coutts, S., Kolotelo, P., Prosser, T., Crosbie, N., Grant, T., Cottam, D., O'Brien, P., and Deletic, A.: Into the deep: evaluation of SourceTracker for assessment of faecal contamination of coastal waters, Water Res., 93, 242–253, https://doi.org/10.1016/j.watres.2016.02.029, 2016.
Igbinosa, I. H., Nwodo, U. U., Sosa, A., Tom, M., and Okoh, A. I.: Commensal Pseudomonas species isolated from wastewater and freshwater milieus in the Eastern Cape Province, South Africa, as reservoir of antibiotic resistant determinants, Int. J. Environ. Res. Public Health, 9, 2537–2549, https://doi.org/10.3390/ijerph9072537, 2012.
Johnson, J. S., Spakowicz, D. J., Hong, B. Y., Petersen, L. M., Demkowicz, P., Chen, L., Leopold, S. R., Hanson, B. M., Agresta, H. O., Gerstein, M., Sodergren, E., and Weinstock, G. M.: Evaluation of 16S rRNA gene sequencing for species and strain-level microbiome analysis. Nat. Commun., 10, 5029, https://doi.org/10.1038/s41467-019-13036-1, 2019.
Karstens, L., Asquith, M., Davin, S., Fair, D., Thomas Gregory, W., Wolfe, A. J., Braun, J., and McWeeney, S.: Controlling for contaminants in low-biomass 16S rRNA gene sequencing experiments, mSystems 4, e00290-19, https://doi.org/10.1128/mSystems.00290-19, 2019.
Knights, D., Kuczynski, J., Charlson, E. S., Zaneveld, J., Mozer, M. C., Collman, R. G., Bushman, F. D., Knight, R., and Kelley, S. T.: Bayesian community-wide culture-independent microbial source tracking, Nat. Methods, 8, 761, https://doi.org/10.1038/nmeth.1650, 2011.
Konrad, C. P. and Booth, D. B.: Hydrologic changes in urban streams and their ecological significance, Am. Fish. S. S., 47, 157–177, 2005.
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D.: Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform, Appl. Environ. Microbiol., 79, 5112–5120, https://doi.org/10.1128/AEM.01043-13, 2013.
Kristian Stevik, T., Kari, A., Ausland, G., and Fredrik Hanssen, J.: Retention and removal of pathogenic bacteria in wastewater percolating through porous media: a review, Water Res., 38, 1355–1367, https://doi.org/10.1016/j.watres.2003.12.024, 2004.
Krone, R. B., Orlob, G. T., and Hodgkinson, C.: Movement of coliform bacteria through porous media, Sewage Ind. Wastes, 30, 1–13, 1958.
Kwon, S. W., Kim, J. S., Park, I. C., Yoon, S. H., Park, D. H., Lim, C. K., and Go, S. J.: Pseudomonas koreensis sp. nov., Pseudomonas umsongensis sp. nov. and Pseudomonas jinjuensis sp. nov., novel species from farm soils in Korea, Int. J. Syst. Evol. Microbiol., 53, 21–27, https://doi.org/10.1099/ijs.0.02326-0, 2003.
Lapworth, D. J., Baran, N., Stuart, M. E., and Ward, R. S.: Emerging organic contaminants in groundwater: A review of sources, fate and occurrence, Environ. Pollut. 163, 287–303, https://doi.org/10.1016/j.envpol.2011.12.034, 2012.
Lassabatere, L., Angulo-Jaramillo, R., Soria Ugalde, J. M., Cuenca, R., Braud, I., and Haverkamp, R.: Beerkan estimation of soil transfer parameters through infiltration experiments – BEST. Soil Sci. Soc. Am. J., 70, 521–532, https://doi.org/10.2136/sssaj2005.0026, 2006.
Le Boulch, M., Déhais, P., Combes, S., and Pascal, G.: The MACADAM database: a MetAboliC pAthways DAtabase for Microbial taxonomic groups for mining potential metabolic capacities of archaeal and bacterial taxonomic groups, Database 2019, https://doi.org/10.1093/database/baz049, 2019.
Le Coustumer, S. and Barraud, S.: Long-term hydraulic and pollution retention performance of infiltration systems, Water Sci. Technol., 55, 235–243, https://doi.org/10.2166/wst.2007.114, 2007.
Lladó, S. and Baldrian, P.: Community-level physiological profiling analyses show potential to identify the copiotrophic bacteria present in soil environments, PLoS One, 12, e0171638, https://doi.org/10.1371/journal.pone.0171638, 2017.
Louca, S., Parfrey, L. W., and Doebeli, M.: Decoupling function and taxonomy in the global ocean microbiome, Science, 353, 1272–1277, https://doi.org/10.1126/science.aaf4507, 2016.
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., and Knight, R.: UniFrac: an effective distance metric for microbial community comparison, ISME J., 5, 169–172, https://doi.org/10.1038/ismej.2010.133, 2011.
Luczkiewicz, A., Kotlarska, E., Artichowicz, W., Tarasewicz, K., and Fudala-Ksiazek, S.: Antimicrobial resistance of Pseudomonas spp. isolated from wastewater and wastewater-impacted marine coastal zone, Environ. Sci. Pollut. Res., 22, 19823–19834, https://doi.org/10.1007/s11356-015-5098-y, 2015.
Lukasik, P., Newton, J. A., Sanders, J. G., Hu, Y., Moreau, C. S., Kronauer, D. J. C., O'Donnell, S., Koga, R., and Russell, J. A.: The structured diversity of specialized gut symbionts of the New World army ants, Mol. Ecol., 26, 3808–3825, https://doi.org/10.1111/mec.14140, 2017.
Mansour, I., Heppell, C. M., Ryo, M., and Rillig, M. C.: Application of the microbial community coalescence concept to riverine networks: Riverine microbial community coalescence, Biol. Rev., 93, 1832–1845, https://doi.org/10.1111/brv.12422, 2018.
Manz, W., Wendt-Potthoff, K., Neu, T. R., Szewzyk, U., and Lawrence, J. R.: Phylogenetic composition, spatial structure, and dynamics of lotic bacterial biofilms investigated by fluorescent in situ hybridization and confocal laser scanning microscopy, Microb. Ecol., 37, 225–237, https://doi.org/10.1007/s002489900148, 1999.
Marsalek, J. and Chocat, B.: International report: Stormwater management, Water Sci. Technol. 46, 1–17, 2002.
Marti, R., Bécouze-Lareure, C., Ribun, S., Marjolet, L., Bernardin Souibgui, C., Aubin, J.-B., Lipeme Kouyi, G., Wiest, L., Blaha, D., and Cournoyer, B.: Bacteriome genetic structures of urban deposits are indicative of their origin and impacted by chemical pollutants, Sci. Rep., 7, 13219, https://doi.org/10.1038/s41598-017-13594-8, 2017.
McCarthy, D. T., Jovanovic, D., Lintern, A., Teakle, I., Barnes, M., Deletic, A., Coleman, R., Rooney, G., Prosser, T., and Coutts, S.: Source tracking using microbial community fingerprints: method comparison with hydrodynamic modelling, Water Res., 109, 253–265, https://doi.org/10.1016/j.watres.2016.11.043, 2017.
McGrane, S. J.: Impacts of urbanisation on hydrological and water quality dynamics, and urban water management: a review, Hydrol. Sci. J., 61, 2295–2311, https://doi.org/10.1080/02626667.2015.1128084, 2016.
Mejía, A. I. and Moglen, G. E.: Spatial patterns of urban development from optimization of flood peaks and imperviousness-based measures, J. Hydrol. Eng., 14, 416–424, https://doi.org/10.1061/(ASCE)1084-0699(2009)14:4(416), 2009.
Mermillod-Blondin, F., Simon, L., Maazouzi, C., Foulquier, A., Delolme, C., and Marmonier, P.: Dynamics of dissolved organic carbon (DOC) through stormwater basins designed for groundwater recharge in urban area: assessment of retention efficiency, Water Res., 81, 27–37, https://doi.org/10.1016/j.watres.2015.05.031, 2015.
Mermillod-Blondin, F., Voisin, J., Marjolet, L., Marmonier, P., and Cournoyer, B.: Clay beads as artificial trapping matrices for monitoring bacterial distribution among urban stormwater infiltration systems and their connected aquifers, Environ. Monit. Assess., 191, 58, https://doi.org/10.1007/s10661-019-7190-0, 2019.
Miyahara, M., Kim, S.-W., Fushinobu, S., Takaki, K., Yamada, T., Watanabe, A., Miyauchi, K., Endo, G., Wakagi, T., and Shoun, H.: Potential of aerobic denitrification by Pseudomonas stutzeri TR2 to reduce nitrous oxide emissions from wastewater treatment plants, Appl. Environ. Microbiol., 76, 4619–4625, https://doi.org/10.1128/AEM.01983-09, 2010.
Murphy, E. M. and Ginn, T. R.: Modeling microbial processes in porous media, Hydrogeol. J., 8, 142–158, https://doi.org/10.1007/s100409900043, 2000.
Noble, R. C. and Overman, S. B.: Pseudomonas stutzeri infection. A review of hospital isolates and a review of the literature, Diagn. Microbiol. Infect. Dis., 19, 51–56, https://doi.org/10.1016/0732-8893(94)90051-5, 1994.
Oksanen, J., Blanchet, F. G., Kindt, R., Legendre, P., Minchin, P. R., O'Hara, R. B., Simpson, G. L., Solymos, P., Stevens, M. H. H., and Wagner, H.: vegan: Community Ecology Package, R package version 2.3–0, available at: https://cran.r-project.org/ (last access: May 2020), 2015.
Park, J.-W. and Crowley, D. E.: Dynamic changes in nahAc gene copy numbers during degradation of naphthalene in PAH-contaminated soils, Appl. Microbiol. Biotechnol., 72, 1322–1329, https://doi.org/10.1007/s00253-006-0423-5, 2006.
Pigneret, M., Mermillod-Blondin, F., Volatier, L., Romestaing, C., Maire, E., Adrien, J., Guillard, L., Roussel, D., and Hervant, F.: Urban pollution of sediments: Impact on the physiology and burrowing activity of tubificid worms and consequences on biogeochemical processes, Sci. Total Environ., 568, 196–207, https://doi.org/10.1016/j.scitotenv.2016.05.174, 2016.
Pinasseau, L., Wiest, L., Fildier, A., Volatier, L., Fones, G. R., Mills, G. A., Mermillod-Blondin, F., and Vulliet E.: Use of passive sampling and high resolution mass spectrometry using a suspect screening approach to characterise emerging pollutants in contaminated groundwater and runoff, Sci. Total Environ., 672, 253–263. https://doi.org/10.1016/j.scitotenv.2019.03.489, 2019.
Pitt, R., Clark, S., and Field, R.: Groundwater contamination potential from stormwater infiltration practices, Urban Water, 1, 217–236, https://doi.org/10.1016/S1462-0758(99)00014-X, 1999.
Pohlon, E., Marxsen, J., and Küsel, K.: Pioneering bacterial and algal communities and potential extracellular enzyme activities of stream biofilms, FEMS Microbiol. Ecol., 71, 364–373, https://doi.org/10.1111/j.1574-6941.2009.00817.x, 2010.
Price, M. N., Dehal, P. S., and Arkin, A. P.: FastTree: computing large minimum evolution trees with profiles instead of a distance matrix, Mol. Biol. Evol., 26, 1641–1650, https://doi.org/10.1093/molbev/msp077, 2009.
Ritchie, M. E., Phipson, B., Wu, D., Hu, Y., Law, C. W., Shi, W., and Smyth, G. K.: limma powers differential expression analyses for RNA-sequencing and microarray studies, Nucleic Acids Res., 43, e47–e47, https://doi.org/10.1093/nar/gkv007, 2015.
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., Lesniewski, R. A., Oakley, B. B., Parks, D. H., Robinson, C. J., Sahl, J. W., Stres, B., Thallinger, G. G., Horn, D. J. V., and Weber, C. F.: Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities, Appl. Environ. Microbiol., 75, 7537–7541, https://doi.org/10.1128/AEM.01541-09, 2009.
Sébastian, C., Barraud, S., Ribun, S., Zoropogui, A., Blaha, D., Becouze-Lareure, C., Kouyi, G. L., and Cournoyer, B.: Accumulated sediments in a detention basin: chemical and microbial hazard assessment linked to hydrological processes, Environ. Sci. Pollut. Res., 21, 5367–5378, https://doi.org/10.1007/s11356-013-2397-z, 2014.
Shuster, W. D., Bonta, J., Thurston, H., Warnemuende, E., and Smith, D. R.: Impacts of impervious surface on watershed hydrology: A review, Urban Water J., 2, 263–275, https://doi.org/10.1080/15730620500386529, 2005.
Smith, R. J., Paterson, J. S., Launer, E., Tobe, S. S., Morello, E., Leijs, R., Marri, S., and Mitchell, J. G.: Stygofauna enhance prokaryotic transport in groundwater ecosystems, Sci. Rep., 6, 32738, https://doi.10.1038/srep32738, 2016.
Tao, Y., Zhou, Y., He, X., Hu, X., and Li, D.: Pseudomonas chengduensis sp. nov., isolated from landfill leachate, Int. J. Syst. Evol. Microbiol., 64, 95–100, https://doi.org/10.1099/ijs.0.050294-0, 2014.
Tedoldi, D., Chebbo, G., Pierlot, D., Kovacs, Y., and Gromaire, M.-C.: Impact of runoff infiltration on contaminant accumulation and transport in the soil/filter media of sustainable urban drainage systems: A literature review, Sci. Total Environ., 569–570, 904–926, https://doi.org/10.1016/j.scitotenv.2016.04.215, 2016.
Tikhonov, M.: Community-level cohesion without cooperation, eLife, 5, e15747, https://doi.org/10.7554/eLife.15747, 2016.
Vadstein, O., Attramadal, K. J. K., Bakke, I., and Olsen, Y.: K-Selection as Microbial Community Management Strategy: A Method for Improved Viability of Larvae in Aquaculture, Front. Microbiol., 9, 2730, https://doi.org/10.3389/fmicb.2018.02730, 2018.
van Loosdrecht, M. C., Lyklema, J., Norde, W., and Zehnder, A. J.: Influence of interfaces on microbial activity, Microbiol. Rev., 54, 75–87, 1990.
Vezzaro, L. and Mikkelsen, P. S.: Application of global sensitivity analysis and uncertainty quantification in dynamic modelling of micropollutants in stormwater runoff, Environ. Modell. Softw., 27, 40–51, https://doi.org/10.1016/j.envsoft.2011.09.012, 2012.
Voisin, J., Cournoyer, B., and Mermillod-Blondin, F.: Assessment of artificial substrates for evaluating groundwater microbial quality, Ecol. Indicators, 71, 577–586, https://doi.org/10.1016/j.ecolind.2016.07.035, 2016.
Voisin, J., Cournoyer, B., Vienney, A., and Mermillod-Blondin, F.: Aquifer recharge with stormwater runoff in urban areas: Influence of vadose zone thickness on nutrient and bacterial transfers from the surface of infiltration basins to groundwater, Sci. Total Environ., 637–638, 1496–1507, https://doi.org/10.1016/j.scitotenv.2018.05.094, 2018.
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R.: Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy, Appl. Environ. Microbiol., 73, 5261–5267, https://doi.org/10.1128/AEM.00062-07, 2007.
Wiest, L., Baudot, R., Lafay, F., Bonjour, E., Becouze-Lareure, C., Aubin, J.B., Jame, P., Barraud, S., Kouyi Lipeme, G., Sébastian, C., and Vulliet, E.: Priority substances in accumulated sediments in a stormwater detention basin from an industrial area, Env. Poll., 243, 1669–1678, https://doi.org/10.1016/j.envpol.2018.09.138, 2018.
Winiarski, T.: Fonction filtration d'un ouvrage urbain – conséquence sur la formation d'un anthroposol, French Ministry of Ecological Transition and Solidarity, TEMIS archives, available at: http://temis.documentation.developpement-durable.gouv.fr/docs/Temis/0081/Temis-0081850/21970_A.pdf (last access: May 2020), 2014.
Winiarski, T., Bedell, J.-P., Delolme, C., and Perrodin, Y.: The impact of stormwater on a soil profile in an infiltration basin, Hydrogeol. J., 14, 1244–1251, https://doi.org/10.1007/s10040-006-0073-9, 2006.
Winiarski, T., Lassabatere, L., Angulo-Jaramillo, R., and Goutaland, D.: Characterization of the heterogeneous flow and pollutant transfer in the unsaturated zone in the fluvio-glacial deposit, Procedia Environ. Sci., 19, 955–964, https://doi.org/10.1016/j.proenv.2013.06.105, 2013.