the Creative Commons Attribution 4.0 License.
the Creative Commons Attribution 4.0 License.
 | 20 Oct 2023
| 20 Oct 2023
Technical note: Discrete in situ vapor sampling for subsequent lab-based water stable isotope analysis
Barbara Herbstritt
Benjamin Gralher
Stefan Seeger
Michael Rinderer
Markus Weiler
Methodological advancements have been made in in situ observations of water stable isotopes that have provided valuable insights into ecohydrological processes. The continuous measurement capabilities of laser-based analyzers allow for high temporal resolutions and non-destructive minimally invasive study designs of such in situ approaches. However, isotope analyzers are expensive, heavy, and require shelter and access to electrical power, which impedes many in situ assays. Therefore, we developed a new inexpensive technique to collect discrete water vapor samples in the field via diffusion-tight inflatable bags that can later be analyzed in the lab. In a series of structured experiments, we tested different procedural settings, bag materials, and closure types for diffusion tightness during storage as well as for practical handling during filling and extraction. To facilitate reuse of sampling bags, we present a conditioning procedure using ambient air as primer. In order to validate our method, direct measurements through hydrophobic in situ probes were compared to repeated measurements of vapor sampled with our bags from the same source. All steps are summarized in a detailed standard operating procedure (SOP). This procedure represents the preparation and measurement of calibration and validation vapor standards necessary for processing of unknown field-collected vapor samples in the foreseen application. By performing pertinent calibration procedures, accuracy was better than 0.4 ‰ for δ18O and 1.9 ‰ for δ2H after 1 d of storage. Our technique is particularly suitable when used in combination with minimally invasive water vapor sampling in situ probes that have already been employed for soils and tree xylem. It is an important step towards minimally invasive monitoring of stable isotope distributions and also time series in virtually undisturbed soils and trees without the need to have an analyzer in the field. It is therefore a promising tool for many applications in ecohydrology and meteorology.
- Article
(1530 KB) - Full-text XML
-
Supplement
(642 KB) - BibTeX
- EndNote





Analyses of stable isotope composition of hydrogen and oxygen (δ2H and δ18O) in soils and plant water have proven to be powerful tools and are therefore widely employed in ecology, hydrology, and related disciplines. Stable isotopes of pore water have been used to provide insights into soil evaporation (Zimmermann et al., 1967; Allison, 1982; Allison et al., 1983; Barnes and Allison, 1988; Walker et al., 1988) and groundwater recharge rates (Dincer et al., 1974; Saxena, 1984; Darling and Bath, 1988; Koeniger et al., 2016). They were used in soil hydrology to study unsaturated and saturated subsurface flow processes and mixing and residence times (Sklash and Farvolden, 1979; Buttle and Sami, 1990; McDonnell, 1990; Stewart and McDonnell, 1991; Gazis and Feng, 2004; Laudon et al., 2004; Garvelmann et al., 2012; Beyer et al., 2016), as well as to quantify evapotranspiration partitioning (Brunel et al., 1997; Hsieh et al., 1998; Yepez et al., 2005; Rothfuss et al., 2010; Wang and Yakir, 2000; Dubbert et al., 2013; Quade et al., 2019). Applications of water stable isotopes in ecology have allowed researchers to identify plant water sources (Dawson and Ehleringer, 1991), to describe water use patterns (Schwinning et al., 2002), and to determine competitive interactions (Ehleringer et al., 1991; Meißner et al., 2012). In plant physiology, insights into plant hydraulic architecture (Drake and Franks, 2003) were possible with isotope techniques, and root water uptake was quantified (Rothfuss and Javaux, 2017; Fan et al., 2017; Seeger and Weiler, 2021), as well as hydraulic lift (Caldwell and Richards, 1989; Meunier et al., 2018).
Conventionally, measurements of pore water and tree xylem water isotope composition are obtained through destructive sampling of soil cores or manual collection of sapwood and subsequent water extraction for isotope ratio mass spectrometry (IRMS) analysis (Ehleringer et al., 2000; West et al., 2006) or isotope ratio infrared spectrometry (IRIS) (Baer et al., 2002; Gupta et al., 2009). These techniques allow for high measurement precision (Horita and Kendall, 2004), but they are comparably expensive in the case of IRMS and generally require highly time-consuming and laborious sample pre-treatment (Kerstel and Gianfrani, 2008). A less expensive and overall more convenient approach, relying on laser-based water stable isotope analyzers, is direct vapor equilibration laser spectrometry (DVE-LS), where samples of soil matrix, rocks, or plant tissue are in equilibrium with a corresponding vapor phase (Wassenaar et al., 2008; Hendry et al., 2015; Gralher et al., 2021).
Although promising, the disadvantage is still that destructive soil sampling or harvesting of plant material generally prevents repeated samples from the exact same position. Additionally, repeated sampling of xylem imposes the risk of killing the tree or weakening it due to fungal infestation. Moreover, taking branch samples can be challenging or even impossible for tall trees. Generally, destructive sampling restricts the number of samples that can be obtained over time and space. This makes high-frequency or even continuous measurements difficult to sustain or simply infeasible. The number and spatiotemporal scope of lab-scale experimental setups as well as environmental isotope studies have continued to expand but are still limited by the available indirect observational techniques (West et al., 2010).
The growing distribution of laser-based water stable isotope analyzers in recent years has also enabled minimally invasive, direct, continuous, and simultaneous measurements of δ2H and δ18O of water vapor. Only from then have time series observations from the same point become possible. Available IRIS instruments allow for measurements at a precision and accuracy comparable to that of IRMS (Berden et al., 2000; Baer et al., 2002; Crosson, 2008; Kerstel and Gianfrani, 2008; Gupta et al., 2009). Importantly, laser-based instruments are portable and therefore potentially field-deployable. The small measurement cavity size (35 mL) of wavelength-scanned cavity ring-down spectroscopy (WS-CRDS) instruments makes them especially ideal for lab-scale experimental setups as well as for small sensor designs. The widespread use of laser-based instruments has therefore stimulated recent developments of a number of new in situ methods for direct measurements of water stable isotopes in various fields. Precipitation measurements were carried out via gas-permeable expanded PTFE surgical tubing (Munksgaard et al., 2011). Soil column breakthrough curves (Herbstritt et al., 2012) as well as analyses of precipitation and canopy throughfall in parallel were achieved via small hydrophobic membrane contactors (Herbstritt et al., 2019). The isotopic composition of pore water was analyzed in lab-scale experiments via hydrophobic microporous tubing (Rothfuss et al., 2013) as well as in natural soil profiles with custom-made hydrophobic porous in situ water isotope probes (WIPs) (Volkmann and Weiler, 2014). Similar in situ probes were also used in tree stems in labeling experiments to analyze the isotopic composition of xylem sap (Volkmann et al., 2016a; Seeger and Weiler, 2021). The “stem borehole method” (Marshall et al., 2020; Kühnhammer et al., 2022) is an alternative way to obtain in situ samples of tree xylem water vapor. Measurements of the isotopic composition of transpired water were conducted using leaf chambers (Wang et al., 2012; Dubbert et al., 2014) or whole-plant chambers (Volkmann et al., 2016b).
The isotopic composition of the liquid water of interest in all these in situ studies was inferred from sampling and measuring a corresponding vapor phase. The water vapor of interest was either withdrawn directly (e.g., from soil profiles or out of tree boreholes) or exchanged and equilibrated with a carrier gas through different types of hydrophobic membranes.
However, operating laser-based analyzers at the study site requires power-consuming, heavy, and expensive equipment to be brought to the field site with the risk of damage and the disadvantage of relative immobility. Consequently, rough or remote terrains, as well as spacious experimental design, exceeding possible tubing lengths between in situ measurements and the isotope analyzer, are virtually excluded with this approach. First attempts to overcome these obstacles by collecting discrete vapor samples under such circumstances into glass bottles were recently presented. In both approaches (see below), the sampled vapor had to be diluted continuously during measurement to compensate for negative pressure when sucked into the analyzer by releasing dry air into the rigid, fragile sampling flasks. The first approach (Havranek et al., 2020) can be seen as a proof of concept. In a follow-up, Havranek et al. (2023) describe a field application of their setup which requires considerable financial resources for the components in use, extensive technical know-how for construction, and substantial effort for field installation of a limited number of flasks. Their recommended operating procedure requires long flushing times, leading to filling times per flask of more than 1 h, which strongly reduces the sample throughput and thus the achievable temporal resolution. The second approach (Magh et al., 2022) partly resolved these issues but still relies on specialty tools, lacks reproducibility due to the small volume of sampled vapor, and currently does not provide the data accuracy needed for natural abundance isotope assays. Further, a sophisticated calculation procedure is necessary for both approaches (Havranek et al., 2020; Magh et al., 2022) to remove the effects of the initial pulse of water vapor during the start of the measurement phase. This initial pulse is mixed with pre-sample vapor, which clearly biases the obtained isotope data.
Therefore, the aim of this study is to develop a technique to collect discrete vapor samples in the field for subsequent lab-based analyses that overcomes the aforementioned problems while still ensuring handiness as well as cost and time efficiency (see Fig. 1). Specifically, we identified a way to prepare and measure calibration and validation vapor standards, necessary for processing unknown field-collected vapor samples. For this purpose, we varied the applied gas flow rate through established non-destructive in situ water isotope probes (WIPs) to improve per-sample time consumption. We tested the diffusion tightness and inertness of various commercially available gas sampling bags as well as custom-made inflatable containers comprising different materials and closing mechanisms. We identified the best performing bags and coupled them with WIPs in order to collect and temporarily store discrete vapor samples prior to lab-based isotope analyses. Also, we identified necessary preparatory steps to optimize the reproduction of in situ data.
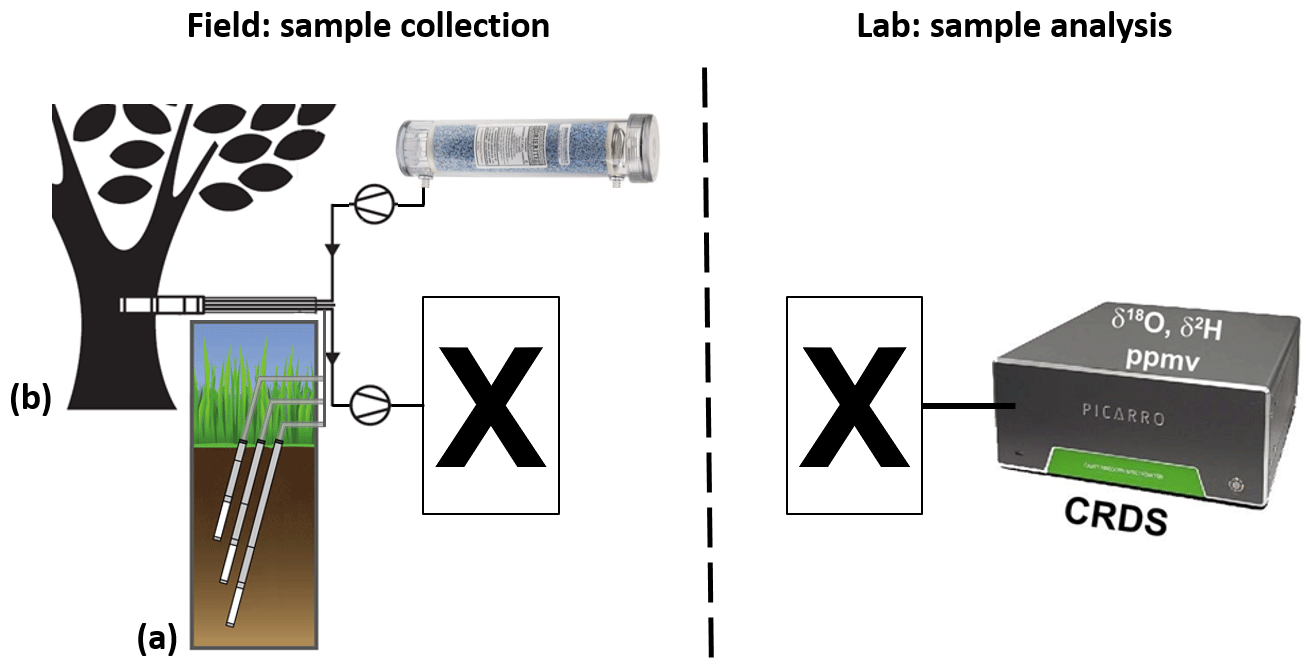
Figure 1Schematic of projected vapor sampling via in situ (a) soil- and (b) xylem-water isotope probes (modified from Volkmann and Weiler, 2014 and Volkmann et al., 2016a). The left part describes the intended field setting, while the right part describes the intended laboratory setting: sampled vapor is first filled into appropriate to-be-identified containers (X) and later analyzed in the lab via CRDS.
2.1 Effect of changing gas throughflow rates on the isotopic composition
In the first part of this study we tried to optimize sample filling times. For this purpose, we investigated the effect of varying gas flow rates through the to-be-employed in situ water isotope probes (WIPs). Originally, the size of the probes was optimized, i.e., the contact area of the membranous tip, to facilitate isotopic equilibrium when the applied flow rates had been set to match the analyzer's demand. This analyzer-immanent prerequisite becomes obsolete for the collection of discrete vapor samples. In the case of discrete vapor sampling through WIPs into airtight containers, any other flow rate can be selected, which inversely affects the filling times of the containers. It is important that the contact area relevant for vapor collection is consistent for all sample collections, as isotope equilibrium cannot be expected anymore when the applied gas flow rates exceed the ones originally recommended (Volkmann and Weiler, 2014). For repeated analysis of a single vapor sample, we found a total gas sample volume of 0.5–1 L to be sufficient (Gralher et al., 2021). Further, we aimed for sampling times in the field of no more than 5 min per sample. Therefore, flow rates of both dry air through the WIP and the corresponding gas sampling rate into the containers were increased stepwise in a lab experiment up to 150 mL min−1 which would yield a 0.75 L sample volume after a filling time of 5 min. We also tested the effects of omitting the originally proposed dilution (Volkmann and Weiler, 2014) when increasing the throughflow rates. In order to keep the gas flow inside the WIP balanced between the inflowing carrier gas and the outflowing sampling gas (and in doing so avoiding overpressure or under-pressure), the sampling rates had to match the throughflow rates. For these tests, a WIP was installed in an evaporation-shielded box filled with moist sand with a water stable isotopic composition of −9.64 ‰ and −66.84 ‰ for δ18O and δ2H, respectively, referenced to the VSMOW-SLAP scale (Craig, 1961a) and kept at a constant temperature of 20.8 ∘C. For precise isotope measurements, we used dry synthetic air (zero air) as carrier gas.
Precise flow rates of the synthetic air from the pressurized gas bottle into the WIP were facilitated by a digital mass flow controller (PN 35828, Analyt-MTC, Müllheim, Germany), while sampling at the same flow rate was facilitated by a small air pump (PN LP27-12, Pollin Electronic GmbH, Pförring, Germany) where the pumping rate can be controlled manually via the applied voltage and controlled with a mass flow meter (PN 35808, Analyt-MTC). The fractions of sampling rates exceeding the analyzer demand of ∼ 35 mL min−1 were vented to air through an open split near the sample inlet port of the isotope analyzer (L2120-i or L2130-i, Picarro Inc., Santa Clara, CA, USA). The analyzer provided quasi-continuous (0.5 Hz) readings of water vapor mixing ratio (in ppmv), oxygen, and hydrogen isotope readings (in ‰), as well as the spectral parameters “h2o_vy”, indicating mixing with ambient air, and “organic_MeOHampl”, indicating spectral interference from the bag material, which we also collected from room air on every day of bag measurements. This setup allowed for facilitating the demanded low and constant stream of gas to the analyzer while at the same time arbitrarily varying the gas flow through the attached WIP.
2.2 Material selection
2.2.1 Diffusivity and spectral interference
Rigid glass or steel bottles and cylinders conventionally used for gas sampling were excluded in our approach, since constant flow through the WIPs was needed in our setup and under-pressure at the analyzer during measurements has to be avoided. Commercially available gasbags with large volumes (10 L) are available with reusable gas valves in opposite to bags with smaller volumes (0.5–2.5 L), which come with septa or other degrading closures. Hence, none of these combinations were suitable for our purposes, either due to their size or due to the type of closure. Other readily available gas sampling bags made of PTFE or laminated aluminum foil from a different supplier were available in appropriate sizes but were available only in minimum order quantities of 10. Their per-order costs ranged between EUR 200 and EUR 350, depending on bag material, which were prohibitive for our purposes. Therefore, we investigated three different inflatable bags of different materials and reasonable sizes, and we combined them with different reclosable caps and valves (Table 1). Additionally, a 2.5 L commercially available gasbag was equipped with a stainless-steel screw-lock valve and also tested. Specifically, we evaluated whether they were sufficiently reliable in terms of diffusion tightness and chemical inertness, while also focusing on easy handling. The criteria for identifying reliable bags or bag material are summarized in a best-practice protocol (standard operating procedure – SOP) in Table A1 in Appendix A of this paper.
Table 1Bag and seal combinations and their properties tested for vapor sampling.

The different bag types were filled with pure N2 (purity 99.996 %) and analyzed immediately thereafter with a CRDS isotope analyzer (L2130-i, Picarro Inc., Santa Clara, CA, USA). We tested PE spout bags (code PE-Sp, PN 1055) and single-layer metallized spout bags (code Al-Sp, PN 1050, both available from Daklapack Europe, Oberhausen, Germany), of which we replaced the original PE spout caps with caps with rubber septa. Filling and continuous vapor isotope analyses were facilitated through these septa via an infusion needle (i.d. = 1 mm) attached to a in. perfluoroalkoxy alkane (PFA) tube. We also tested two kinds of three-layer metallized zip bags with fill volumes of 1 L (PN: CB400-420BRZ, color: red) and 0.5 L (PN: CB400-310GZ, color: gold, both available from Weber Packaging GmbH, Güglingen, Germany). They were heat-sealed and equipped with silicone blots on the outside, which served as septa after 2 d of drying (code Al3-Sil for the 1 L bag, code Al3g-Sil for the 0.5 L bag). The bags were filled, and their content withdrawn for isotope analysis through the silicone septa again via an infusion needle attached to a in. PFA tube.
To improve handiness and simplify filling and sample analysis when using the three-layer metallized bags, we tested two different types of valves as alternatives to our custom-made silicone septa. With a punching tool, a hole was applied to each bag for the respective diameter of the screw connections. We fixed small pneumatic brass couplings (PN KDG M5 NW2,7; Landefeld, Kassel, Germany) on the three-layer metallized 1 L zip bags (code Al3-PC), which were then heat-sealed. The respective plug connector (PN KSGI M5 NW2,7; Landefeld, Kassel, Germany) was connected to a in. PFA tube for filling and analysis. Also, stainless-steel screw-lock gasbag valves with 6 mm hose fittings (PN 11701150, Linde, Pullach, Germany) were mounted onto 0.5 L and 1 L three-layer metallized zip bags (codes Al3s-GbV and Al3-GbV) after punching a 10 mm hole. For increased gastightness, we mounted an additional custom-made rubber washer between the valve and the inner wall of the bags which were then heat-sealed. For filling as well as sample analysis, the 6 mm hose fitting was adapted to a in. PFA tube.
To detect gradual diffusive exchange of the bag content with ambient air or outgassing from the employed material, three to five replicates of each bag–valve combination were flushed with N2 and evacuated twice before they were again filled with pure N2. They were then stored at ambient temperature and repeatedly analyzed over the course of 4 weeks. Measurement frequency was every 2 to 3 d during the first 2 weeks and one final time at the end of the fourth week, unless a bag–valve combination was found unsatisfactory earlier.
2.2.2 “Climate chamber” experiment
To further test the vulnerability of projected discrete vapor samples, i.e., to test if relative humidity outside of the bags can exchange with and thus flaw the sample inside the bag, we designed a small climate chamber which consisted of a plastic box (inner dimensions 57 cm × 37 cm × 32 cm) covered by a plastic lid with all holes and slits taped. We prepared six Al3s-GbV bags filled with vapor from the same source (described in detail in Sect. 2.3). Three bags were placed in the box directly, while the other three were inserted in metal cans of the bags' size prior to placement in the box. Such metal cans are normally used for the transport and storage of glass bottles containing liquid water sampled for dissolved gas analysis. They are considered diffusion-tight when closed and sealed by means of metal lids, rubber seal rings, and metal clasps, as we did. Inside the box, we also placed an open bowl of water (ca. 350 mL) to quickly reach and then maintain a relative humidity near 100 % over the course of the test which lasted 3 weeks. The box was deposited in the basement of our laboratory building to facilitate fairly stable temperature conditions. Temperature (∘C) and relative humidity (%) inside the box were recorded every 10 min with a CS215 probe connected to a CR200 logger (both from Campbell Scientific, Logan, UT, USA). These data were then converted to water vapor mixing ratios using Magnus' equation (Foken, 2008). Vapor concentration (ppmv) and isotope (δ18O, δ2H) data from the vapor source used for filling the bags, from the inside of the box after 20 d, and from all canned and un-canned bags after 20 d of storage inside the box were collected with a Picarro 2120-i.
2.3 Field trial
We tested the reliability of the projected sampling procedure first in a laboratory experiment and later in the field using Al3-GbV bags. For this purpose, four evaporation-shielded boxes (V= 18 L), with moist sands with similar water contents but different water isotopic compositions were prepared, and a WIP (Volkmann and Weiler, 2014) was installed in each of the boxes to sample their soil water vapor. We used a low-weight sampling setup that provided a constant air flow of 150 mL min−1 with the small pumps described above (Sect. 2.1). The incoming stream of ambient air was dried by a Drierite drying column (PN 26800, W. A. Hammond Drierite Co. Ltd., Xenia, OH 45385, USA) and directed through the throughflow line into the porous tip of the WIP. The vapor generated inside the WIP was withdrawn through the sampling line by a second small pump with the same gas flow rate. The stream of sampled vapor was directed to the isotope analyzer (demanding 35 mL min−1), using an open split for excess vapor (115 mL min−1) near the analyzer's sample inlet port. Immediately after the direct measurement, the analyzer was disconnected, and the setup was used for directing the entire gas stream into the bags. Additionally, ambient vapor data were collected with the isotope analyzer. Replicates of sampled vapor were filled in bags, which were then analyzed 2 h later. Calibration was facilitated using in situ and bag measurements of those samples displaying the highest and the lowest δ2H values, treating these in situ values de facto as standards. This selection was maintained for calibration of δ18O values, too, although that meant that the so-selected standards did not “bracket” the to-be-calibrated “samples” as is common best practice. The precision would be the standard deviation of repeated calibrated isotope readings of the samples displaying intermediate δ2H readings. The accuracy would be the deviation of the calibrated mean of replicates of the respective in situ measurements. Note that this led to the reproduction (validation) of the intermediate in situ values rather than liquid water values referenced to the VSMOW-SLAP scale (Craig, 1961a). The derivation of liquid water values from vapor isotope observations has been described in Volkmann and Weiler (2014).
2.4 Reusability of sampling bags
2.4.1 Flushing attempts
To test the reusability of the bags, we applied the following flushing procedure. Al3-GbV and Al3s-GbV bags were filled with pure N2, evacuated immediately thereafter with a LABOPORT® diaphragm vacuum pump (N810.3 FT.18, KNF Neuberger GmbH, Munzingen, Germany), and filled again with pure N2. On the next day, they were evacuated again, filled with pure N2 again, and evacuated one final time. After these preparatory steps, three to five bag replicates were used for sampling vapor from sources both isotopically different and identical to the ones that had been sampled before with the respective bags. The setup used for this purpose was identical to the one described in Sect. 2.3 except that different diaphragm gas pumps (BOXER 22K, Boxer, Ottobeuren, Germany) were used. Vapor concentration and isotope data of the bags were recorded with a Picarro L2120-i 1, 3, and 7 d after vapor sampling.
2.4.2 Conditioning
We additionally tested the reusability of the bags by comparing two ways of conditioning previously used sampling bags. The first way was by filling a batch of bags with dry synthetic air, leaving them filled for at least 1 d, analyzing their vapor concentration and isotope signature with a Picarro L2120-i, evacuating them, filling them again, and repeating this cycle several times. The second way was identical except that moist, isotopically homogeneous air was used for filling and priming. In both cases, the absolute vapor concentrations and the standard deviations (SDs) of isotope readings from repeated batch measurements were considered as predictor for conditioning efficiency. Efficiency was then scrutinized by using the so-conditioned bags for collecting vapor samples from isotopically diverse sources (setup details in Sect. 2.3) followed by repeated analyses over the course of up to 7 d. Mean vapor isotopic compositions of these sources were −20.41 ‰, −29.32 ‰, and −37.24 ‰ for δ18O and −84.95 ‰, −139.53 ‰, and −195.77 ‰ for δ2H. Again, calibration was facilitated using bag measurements of those samples with the highest and lowest isotope values and their respective in situ measurements. We report precision and accuracy as quality measures of the calibration process. The reported precision is the SD of the repeated calibrated isotope readings of the intermediate source which we consider as validation standard, while the reported accuracy is the deviation of the calibrated mean of repeated measurements from the respective target value, which are the isotope readings from the respective in situ measurements.
3.1 Effect of changing gas throughflow rates on the isotopic composition
With increasing flow rates through the WIP, the vapor content originating from the source water decreased in the sampling gas (Fig. 2). At the same time, the isotopic composition for both isotope ratios under investigation changed, indicating that no equilibrium between the source water and the provided dry gas stream was established inside the WIP. Such kinetic fractionation effects could be observed at all flow rates exceeding the originally proposed equilibrium flow rate from Volkmann and Weiler (2014). They were stronger for δ18O than for δ2H. The induced decreasing vapor contents were strongly correlated with decreasing isotopic signatures. In the case of the WIP operated with proportional dilution and throughflow rates, vapor content ranged from 10 325 to 14 432 ppmv, while the concurrent uncalibrated isotope readings were in a range of −18.92 ‰ to −30.99 ‰ in the case of δ18O and −136.85 ‰ to −143.41 ‰ in the case of δ2H. The highest values of these ranges correspond to the settings originally proposed by Volkmann and Weiler (2014). These changes correspond to change rates of 0.241 ‰ (mL min−1) for δ18O and 0.131 ‰ (mL min−1) for δ2H. Similar observations were made for the case when dilution was set to zero at higher flow rates. Here, the vapor content ranged from 12 256 to 20 041 ppmv, while the uncalibrated isotope readings varied between −30.35 ‰ and −39.13 ‰ in the case of δ18O and between −143.23 ‰ and −146.95 ‰ in the case of δ2H. Here, the changes correspond to change rates of 0.117 ‰ (mL min−1) for δ18O and 0.049 ‰ (mL min−1) for δ2H. Vapor was always sampled from the same liquid water source which had an isotopic composition of −9.64 ‰ and −66.84 ‰ for δ18O and δ2H, respectively, referenced to the VSMOW-SLAP scale (Craig, 1961a). All coefficients of determination (R2) between vapor content and isotope readings were greater than 0.99. At the target flow rate of 150 mL min−1 throughflow and 0 mL min−1 dilution, vapor content was close to 12 000 ppmv at 20.8 ∘C (Fig. 2).
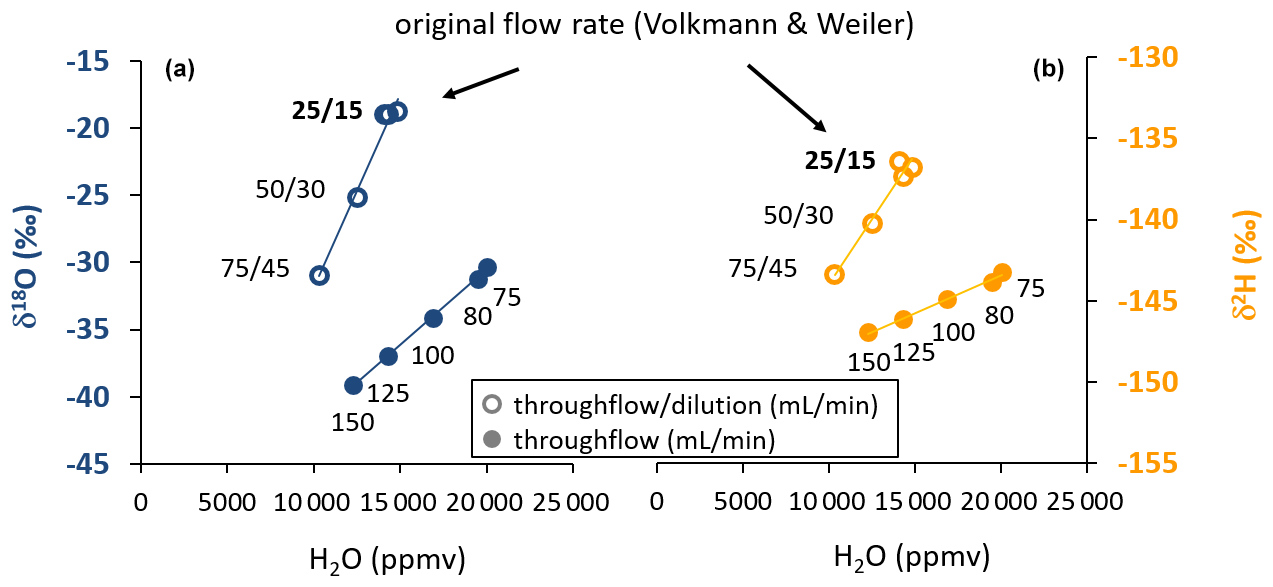
Figure 2Scatterplot of water vapor isotopic composition (a: δ18O, b: δ2H) and vapor content obtained by varying gas throughflow rates with (open symbols) and without (closed symbols) dilution at a constant temperature. This pattern illustrates the kinetic fractionation effects with increasing flow rates through the probe. The numbers refer to the respective gas flow rates applied as throughflow/dilution or throughflow-only during the tests. The flow rates originally proposed by Volkmann and Weiler (2014) facilitated equilibrium according to the design of the probe's tip size and the known instrument flow rate.
3.2 Material selection
3.2.1 Diffusivity and spectral interference
Diffusion tightness and long-term storage effects
During the test for diffusion tightness, the water vapor content readings of the ambient air in the lab were in a range of 9000–18 000 ppmv, while inside the bags they were initially close to zero due to pure N2 inflation. Over time, these vapor pressure gradients were gradually leveled out with different rates. This is qualitatively evident from the different slopes of the dashed lines (Fig. 3). PE spout bags (PE-Sp) displayed the highest increase when after 3 d the vapor content had already increased by ∼ 7000 ppmv. Vapor content readings in the metallized spout bags (Al-Sp) were ∼ 4000 ppmv after 3 d. Three-layer metallized bags displayed the lowest vapor increase rate. Design and thus airtightness of these bags differed only with the type of septum or valves used. The vapor content readings in the bag types with the silicon blot (Al3-Sil) were ∼ 1200 ppmv after 4 d.
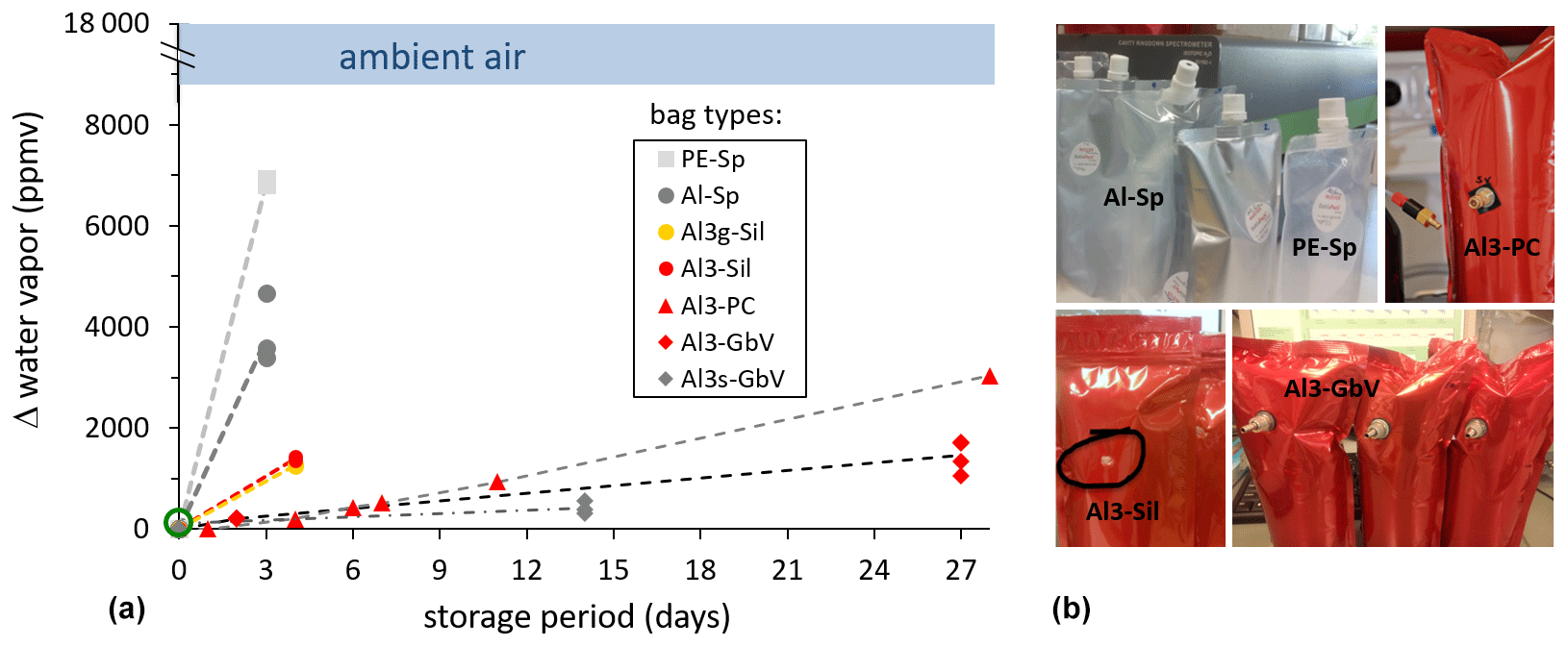
Figure 3(a) Time series of vapor content readings inside different bag types (closed symbols), initially filled with pure N2 (green open circle). (b) Example pictures of tested sampling bags.
Only for the better performing bag–valve combinations (Al3-PC, Al3-GbV, and Al3s-GbV) did we extend the vapor content measurements up to 4 weeks. After this period, mean vapor content readings in Al3-PC and Al3-GbV bags were ∼ 3000 ppmv and ∼ 1450 ppmv, respectively. Mean vapor content readings in Al3s-GbV bags were ∼ 420 ppmv after 2 weeks, which was the final value possible for this bag type due to its smaller volume.
Spectral interference of outgassing material
In two bag types, deviations of the spectral parameters from the pure N2 signal were found, which were also correlated with the respective isotope readings. The spectral line width variable indicative for gas composition (“h2o_vy”) is 0.4309 ± 0.0015 ppm on our L2130-i analyzer for air containing oxygen at atmospheric levels, while it is 0.4563 ± 0.0049 ppm for pure N2. Vapor concentration of bag-type Al3g-Sil containing pure N2 was below 2000 ppmv and should therefore plot at a h2o_vy value of about 0.46 ppm like the Al3-Sil bags (same material but different color) but evolved towards 0.43 ppm. Simultaneously, apparent enrichment in heavy isotopes was observed in Al3g-Sil bags with an increase of around 80 ‰ in δ18O and 150 ‰ in δ2H compared to samples stored in Al3-Sil bags.
In the commercially available 2.5 L Plastigas® bag equipped with a gasbag valve (PN 11701150, Linde) (combination not featured in Fig. 3), we observed an apparent depletion in δ2H from −190 ‰ to −305 ‰ after 24 h and to −360 ‰ after 72 h. At the same time, a spectral variable recorded on the L2120-i analyzer, indicating potential contamination with organic compounds (“organic_MeOHampl”), increased to 0.00760 ± 0.00014 after 24 h and to 0.01005 ± 0.00012 after 72 h. The initial value was 0.00095 ± 0.00026, which was also observed in ambient air. We therefore also excluded Al3g-Sil and Plastigas® bags from further testing due to the observed spectral interferences. Further tests and isotope samplings in our study were conducted with Al3-GbV or Al3-GbV bags only. The performance of the different bag materials in the tests is summarized in Table 2. The protocol for testing bags and material properties, as well as the respective target values for passing the tests, can be found in Appendix A.
Table 2Performance of tested bag types. Test passed is indicated by ✓, failed by X, and not conducted by –.
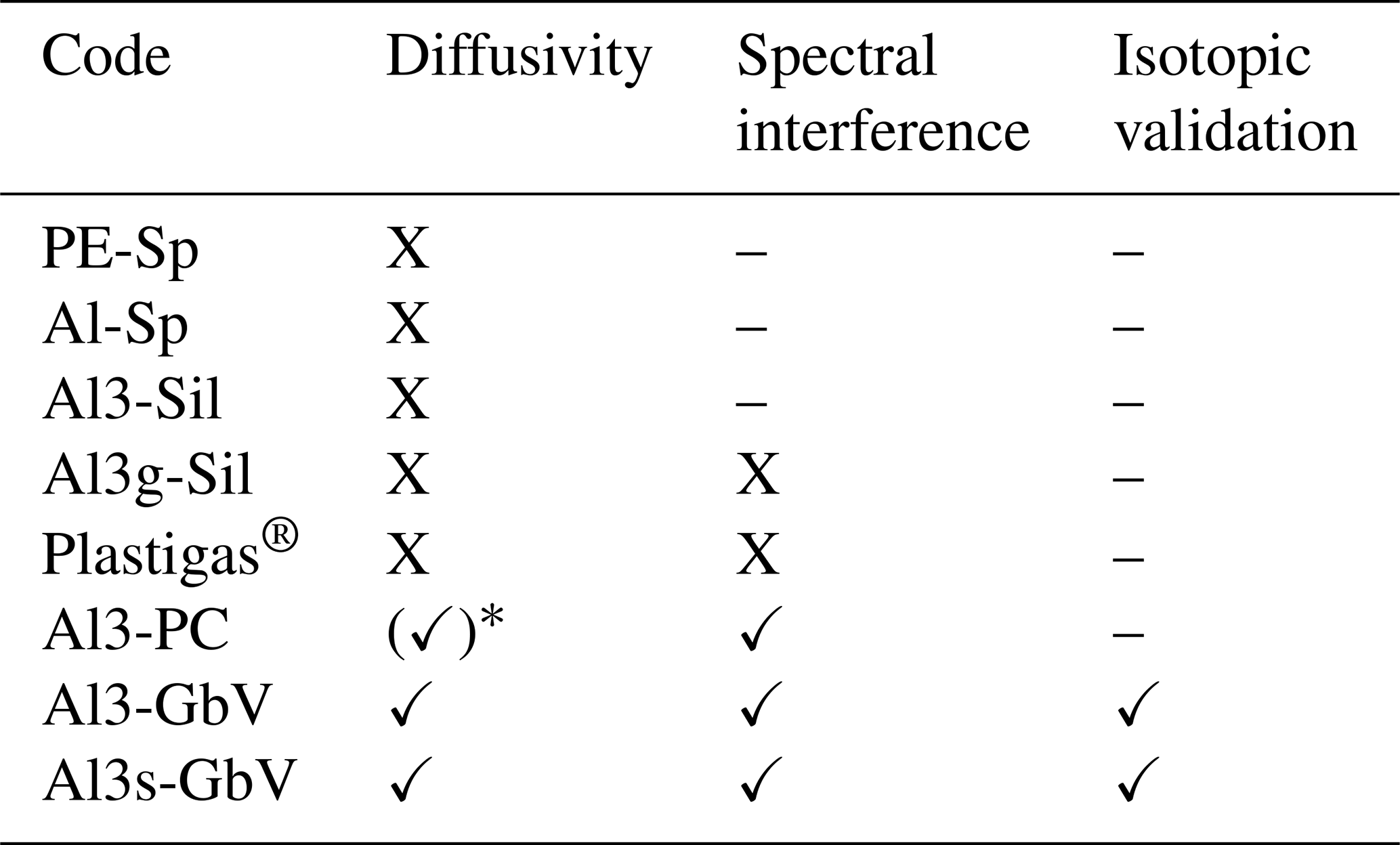
* coupling corrosive
3.2.2 “Climate chamber” experiment
Temperature was quite stable inside the climate chamber, as intended. It ranged between 18.1 and 16.3 ∘C. Relative humidity rose to > 97 % within 6 h and maintained on average at 99.5 % throughout the remaining observation period, which translated to vapor mixing ratios of 19 972 ± 519 ppmv. Mean vapor contents from the source were 14 365 ppmv during filling and 12 574 ppmv inside all bags after 20 d. Differences in mean vapor contents between canned and un-canned bags were smaller than the respective variations within the two batches. Isotope readings of all bags appeared to be enriched by 2.9 ‰ in δ18O and 15.0 ‰ in δ2H relative to the source, again with negligible differences between the two batches (data shown in Fig. S1 in the Supplement).
3.3 Field trial
When comparing all isotope data of the direct in situ sampling from the sand boxes in the field with the data from vapor sampled into the bags and measured 2 h later, we found no systematic bias towards congruent enrichment or depletion in heavy isotopes. Instead, all raw isotope data of the bag measurements appeared to be shifted towards ambient air values by −0.6 ‰ to +1.6 ‰ for δ18O and +4 ‰ to −5 ‰ for δ2H relative to their respective in situ measurements. We used isotope readings of in situ and bag measurements from the samples displaying extreme δ2H values for calibration. Thereby, in situ values of the intermediate samples could be reproduced by calibrated bag measurements with a precision of 0.15 ‰ and an accuracy of 0.8 ‰ for δ18O. The respective numbers for δ2H were 0.76 ‰ and 2.89 ‰ (Fig. 4).
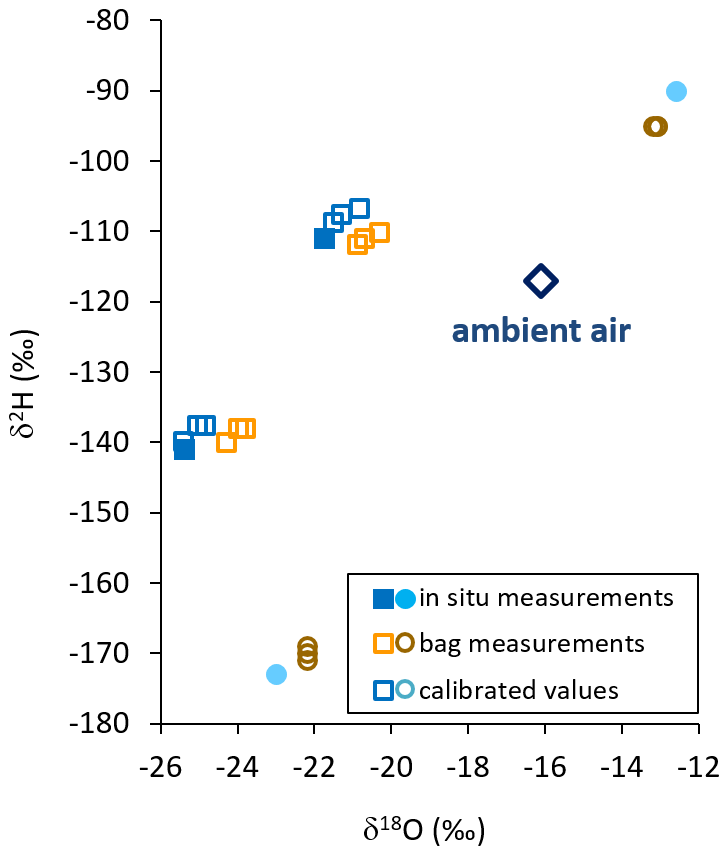
Figure 4Isotope data from in situ measurements (filled symbols), raw bag measurements (orange and brown open symbols), and calibrated in situ data calculated from bag measurements (blue and cyan open symbols) using in situ (cyan closed symbols) and raw values (brown open symbols) of sources displaying extreme δ2H values. Ambient vapor at that day is displayed as a blue open diamond.
3.4 Reusability of sampling bags
3.4.1 Flushing attempts
Generally, isotope data obtained from repeated measurements of the bags unanimously trended towards the values recorded previously from the respective sample bags. These trends increased over time and changes were proportional to the differences between current in situ and previous bag measurements on the individual bag level. Such memory effects persisted although the bags had been evacuated and flushed three times with dry N2 prior to vapor sampling. The trends were consistent for all tested bag types and appeared to be independent of concurrent ambient air values for both isotope ratios investigated (data shown in Fig. B1 in Appendix B).
3.4.2 Conditioning
The two different conditioning methods (dry and moist) applied to previously used sample bags yielded contrary results. Conditioning with dry synthetic air caused vapor content readings to decrease stepwise down to 324 ppmv (Fig. 5a), while isotope signatures became more enriched (isotope data shown in Fig. S2). Their SDs generally decreased but remained above 2.9 ‰ for δ18O and 18.8 ‰ for δ2H (Fig. 5b). In contrast, conditioning with moist air resulted in vapor content readings to decrease to 6740 ppmv, which was in the order of magnitude of the level of conditioning. Isotope signatures of so-primed bags clustered around the conditioning values (data shown in Fig. S3) with SDs decreasing to 0.05 ‰ for δ18O and 1.07 ‰ for δ2H after four to five steps (Fig. 5b).
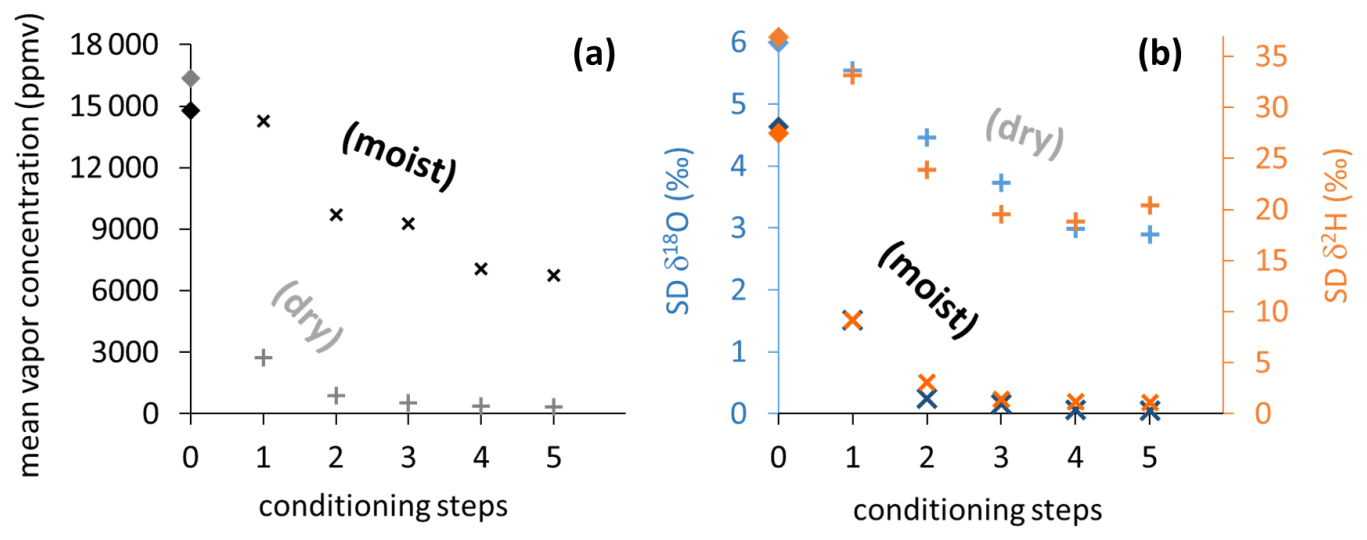
Figure 5Vapor content readings (a) and SDs of isotope readings (b) from vapor sampling bags, stepwise conditioned with dry synthetic air (light plus symbols) and moist air (dark cross symbols). Light and dark diamond symbols on vertical axes represent mean values of pre-conditioning sample measurements.
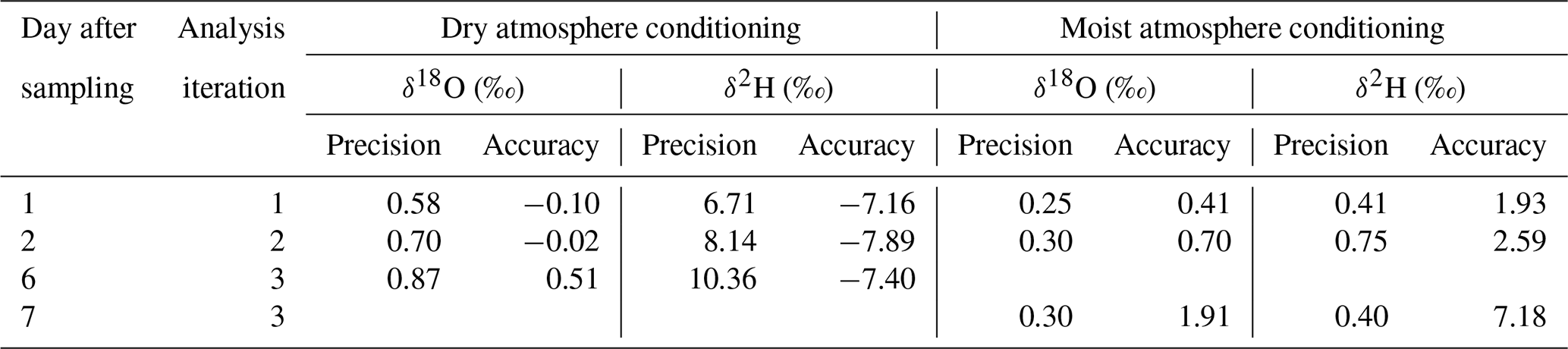
Reusing the conditioned bags for sampling vapor from isotopically diverse sources also yielded contrasting results. SDs of isotope readings in bag replicates after dry conditioning were larger than after moist conditioning. Repeated measurements generally resulted in a decline of measurement precision and accuracy of mean isotope values (Fig. 6). Comparing in situ values with bag measurements yielded no consistent picture in the case of dry conditioning (Fig. 6a), whereas in the case of moist conditioning a bias towards the conditioning values became evident, which increased over time (Fig. 6b). One day after filling, raw isotope data of the bag measurements deviated by −0.6 ‰ to +1.6 ‰ for δ18O and +4 ‰ to −5 ‰ for δ2H relative to their respective in situ measurements. Calibration for reproduction of the intermediate in situ values worked better for moist- than for dry-conditioned bags. This refers to the precision as well as to the accuracy (Table 3).
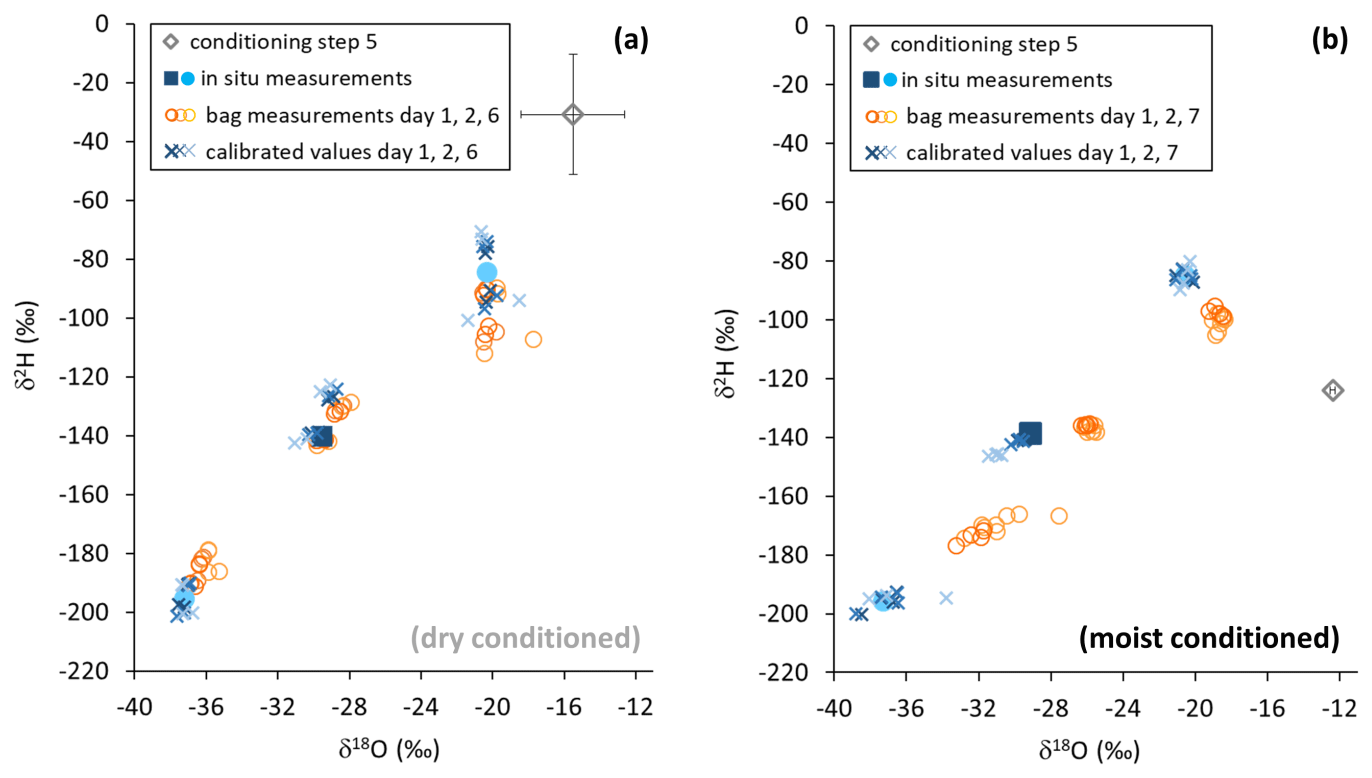
Figure 6Isotope data from in situ measurements (filled symbols), gasbag measurements (open circles), and in situ data calculated from gasbag measurements (asterisks) after conditioning with a dry (a) and moist atmosphere (b). Pre-sampling levels are represented by gray diamonds. Note that error bars (black) are smaller than the symbol in the case of moist conditioning (b).
Table 3Conditioning effects on vapor stable isotope measurements from reused sampling bags.
4.1 Gas flow rate effects
The goal of this study was to facilitate storage of in situ sampled vapor without sensitive and costly analytical equipment in the field. The main applications we had in mind were highly frequent minimally invasive in situ isotope measurements of water vapor in soils and plant tissue as recently performed by Volkmann and Weiler (2014), Volkmann et al. (2016a), Seeger and Weiler (2021), or Gessler et al. (2022). Their setups were based on applying stable yet low gas flow rates to their WIPs. These flow rates resulted in continuous vapor samples assumed to be in isotopic equilibrium with the liquid water of interest. They had been carefully adjusted to the employed analyzers' demands. However, in the intended absence of an analyzer this necessity becomes obsolete. Therefore, we first tested the effect of higher gas flow rates on the obtained vapor isotope signatures. We did so in order to facilitate shorter filling times that would allow for higher filling frequencies of any containers of sufficient volume (0.5–1 L) needed to collect and store vapor samples prior to lab-based analyses.
Incidentally, we also tested the necessity of isotope equilibrium for water vapor sampling using WIPs. We found that also under non-equilibrium conditions in situ isotope values originating from wet sand standards with equal matrix potentials could be reproduced with a very good precision and accuracy. We attribute this to the fact that all employed WIPs had been constructed equal in size. This resulted in consistent gas transit and vapor pickup times inside the tips as well as consistent relevant soil contact area, which is the vapor collection area. This expands the findings of previous studies which aimed at facilitating equilibrium conditions when sampling water vapor from soils or stem boreholes (Volkmann and Weiler, 2014; Marshall et al., 2020). This also means that our approach may not be suitable for application together with the stem borehole method as the tree stem diameters will likely be variable, thus not allowing for consistent gas transit and vapor pickup times. By using identical WIPs throughout our experiment as well as for standards and samples in the foreseen application, potential isotope fractionation effects induced by the membrane are eliminated in the calibration process. Such membrane-induced fractionation effects have been observed before during across-membrane collection of vapor for liquid water isotope determination (Herbstritt et al., 2012). Further, non-equilibrium conditions as encountered in this study did not require any extra correctional efforts. Mathematically, the applied calibration routine is identical to the case of the direct vapor equilibration method (Wassenaar et al., 2008; Hendry et al., 2015; Gralher et al., 2021) or even routine, automated liquid water isotope analyses (Werner and Brand, 2001). Nonetheless, we are aware that differences in matrix potential among samples or relative to the co-prepared standards in the foreseen application may result in differences in the deviation from isotope equilibrium values when applying high flow rates as we did. This would require extra post-processing measures besides our straightforward calibration process. However, such tests were outside the scope of this study. Potentially, if time permits, low flow rates facilitating equilibrium could be a workaround ideally superseding such measures.
Although pure N2 as a carrier gas would have been cheaper, we used synthetic air in order to maintain a consistent nitrogen-to-oxygen mixing ratio. This helped to avoid gas matrix effects previously demonstrated for CRDS instruments (Gralher et al., 2016). We found that gas flow rates higher than the ones previously applied (Volkmann and Weiler, 2014; Volkmann et al., 2016a) immediately resulted in incomplete equilibrium between the liquid water under investigation and the obtained vapor sample (Fig. 2). This became evident by decreasing below-saturation vapor contents as well as lighter isotope readings. On the other hand, we found that even the highest gas flow rates applied in this study (150 mL min−1) still yielded vapor concentrations (∼ 12 000 ppmv at 21 ∘C) that are high enough to be within the analyzer's optimum measurement range and thus enable sufficiently precise isotope measurements for resolving natural variations (Picarro Document Library, 2021). At the same time, a dilution of the obtained vapor stream is obsolete under such settings as the low obtained vapor concentrations impose no risk of condensation. Of course, field sites being sampled for vapor while enduring temperatures that are much higher than the lab temperature might yield vapor concentrations that are too high. Then, even higher flow rates or the reapplication of a dilution flow is necessary for compensation to avoid condensation and thus un-correctable isotope fractionation. This implies that like for other indirect minimally invasive methods (Volkmann and Weiler, 2014; Magh et al., 2022) knowledge about the temperature or maintenance of its consistency at the points of vapor sampling is mandatory for interpreting the obtained isotope data. Cases where temperature differs considerably among sampling sites and/or the site of calibration standard preparation require additional correction schemes considering the temperature dependencies of water-vapor isotope fractionation. This could be facilitated either via mathematical approaches based on the dependencies described by Majoube (1971) or via empirical approaches derived from sets of calibration standards that were collected while intentionally being subjected to different, controlled temperatures. Calibration standards can be prepared by installing WIPs in evaporation-shielded sand-filled boxes wetted with water of known isotopic composition as detailed in Volkmann and Weiler (2014).
The changes in isotope readings and thus deviations from equilibrium were smaller for δ2H than for δ18O in absolute numbers (10.1 ‰ vs. 20.21 ‰, respectively) (Fig. 2) and even more so in relation to naturally occurring isotope variations, where changes usually exhibit an 8 : 1 ratio in meteoric waters (which is the slope of the Global Meteoric Water Line; Craig, 1961b). Therefore, in situ isotope assays relying on discrete vapor sampling for later analysis with a setup similar to the one foreseen here have to ensure precise control of the applied gas flow rates for samples and co-measured standards in order to comply with the paramount “principle of identical treatment” (Werner and Brand, 2001). If the isotope ratio under investigation is optional, we recommend interpreting hydrogen rather than oxygen isotope ratios given the lower susceptibility regarding gas flow rate effects and thus a more favorable signal-to-noise ratio. In this context, it is quite convenient that labeling studies are more cost-efficient when using deuterium as tracer rather than oxygen-18 (Magh et al., 2022).
Flow rates through the probes, exceeding previously recommended settings (Volkmann and Weiler, 2014) immediately resulted in changes of vapor concentration and isotope readings (Fig. 2). Hence, both the previous recommendation and our newly selected settings require precise control of flow rates, as any uncertainty in flow rate settings translates to systematic errors in isotope readings. These errors are considerably higher at lower flow rates and higher for δ18O than for δ2H. Aiming at lower flow rates in order to reliably achieve equilibrium readings appeared impractical in our study for two reasons. Firstly, the employed analyzer's gas flow demand defines the minimum total gas flow. A workaround, applying higher dilution flow rates, would yield varying vapor concentrations and thus vapor concentration effects as can be seen from the different isotope readings observed for the 75 mL throughflow rate with or without dilution (Fig. 2), thus potentially introducing additional errors. And secondly, applying lower flow rates contradicts the study aim of shorter filling times and thus higher sample collection frequencies and achievable temporal resolution.
4.2 Material and closure type selection
Our purpose was to find an inexpensive yet reliable vapor sample container as an alternative to commercially available gasbags, which we found to be insufficient for our purposes either due to their large size or due to their degrading closure type. Therefore, we also inflated other types of sample bags with pure, dry nitrogen gas. We found that only those combining laminated aluminum (Al) foil bodies and metal screw-lock valves yielded useful barriers against ambient vapor pressures (Fig. 3). These bags are mass produced, originally produced for storing food or cosmetics, and are much cheaper than existing diffusion-tight containers from specialized suppliers. All other tested combinations of bag materials and locks failed to prevent intrusion of ambient air, which is crucial for the storage of vapor samples. This corresponds to the finding of a previous study where Al-laminated bags also performed best in avoiding evaporation from soil samples (Gralher et al., 2021). Although the number of water molecules of these samples were 3 magnitudes higher than in our study, we were not surprised to find Al-laminated bags again ranking best in material suitability.
Interestingly, not only the material but also the color of the material coating the diffusive barrier appeared to play a role. Clearly, different colors are a result of different chemical complex formulas used in the production and dyeing process. Unfortunately, they seem to come with different outgassing properties and spectral interference potentials regarding the intended isotope analyses. This issue always needs to be checked in advance. The presence of certain organic compounds in a given gas sample may flaw laser-based isotope readings (Brand et al., 2009; Hendry et al., 2011), some of which can be identified by changes in the spectral parameter readings of the CRDS isotope analyzer.
In the climate chamber experiment, we actually sampled vapor in order to test how its concentration would change when the container is subjected to extreme moisture conditions over a longer time period. Unexpectedly, we found that vapor concentrations slightly decreased over time (Fig. S1). This clearly contradicted the applied vapor concentration gradient relative to ambient conditions. Further, the extent of decrease was not affected by the canning of some of the containers as typically proposed for sampling water for dissolved gas analyses. Therefore, we argue that the employed containers were sufficiently diffusion-tight against ambient meteorological forcings and also that a fraction of the sampled vapor must have been absorbed by the inside coating of the containers (which had been repeatedly flushed with a dry atmosphere prior to vapor sampling). We can only speculate that mixing with previously absorbed vapor led to the observed, fairly uniform enrichment in heavy isotopes for both δ18O and δ2H. Presumably, the increase in vapor concentration inside Al-laminated bags following pure nitrogen gas inflation in the first part of the material tests (Fig. 3) was mainly due to release of previously absorbed vapor rather than via diffusive intrusion from ambient.
4.3 Field trial
This conditioning effect from previous filling or exposures would also explain why isotope data from the field-derived samples unanimously appeared to shift towards ambient air in dual-isotope space (Fig. 4). This was likely because all bags used for this part of our study had been freshly prepared. In doing so, during valve installation they had all been exposed to the same ambient air (in the lab) with a homogeneous vapor isotopic composition. Apparently, that air was isotopically quite similar to the one recorded during the filling of the field samples. The proportional, systematic shift of isotope values allowed for a robust calibration scheme to be applied. This scheme used in situ and bag measurements of those samples displaying extreme δ2H values. It reproduced the intermediate in situ isotope values representative of sampling of the bags with a precision sufficient for resolving naturally occurring variations of meteoric waters. The observed discrepancies between in situ and bag measurements indicate that the principle of identical treatment needs to be followed also in terms of ambient vapor isotopic composition when preparing a batch of sample bags for the collection of unknown samples as well as co-measured standards. Additionally, we recommend using bags of identical size and inflating them to the same volume in order to maintain a uniform ratio of sample volume to internal wall area.
4.4 Reusability of sampling bags
Initially, we had used dry air or pure nitrogen gas in an attempt to erase the signal of previous fillings and thus avoid carryover effects when reusing the sampling bags. However, this procedure did not produce the desired outcome. Repeated measurements from identically filled bags still displayed a clear isotope pattern of the previous fillings (Fig. A2). The range of this pattern must be associated with the measurement uncertainty if the latest filling had been an unknown sample and would thus prevent resolving natural isotope variations. It was established before the first measurement after 5 d and then seemed to persist.
Therefore, we tried to find a different easy-to-implement flushing or conditioning routine that would repeatedly enable precise vapor isotope measurements using our sampling bags. For this purpose, we systematically compared the impact of dry and moist conditioning on the measurement precision and accuracy of vapor from isotopically diverse sources. From experience, we knew that room air vapor isotope signatures are usually quite persistent on shorter timescales as necessary for filling batches of sampling bags using high-flow rate devices such as vacuum pumps. Using this unlimited resource, we were able to establish a conditioning routine that is easy to reproduce and enables the precise reproduction of in situ isotope values following impertinent calibration schemes (Table 3). In this context, the low SD of isotope readings from moist-conditioned bags proved to be a far better predictor of conditioning efficiency than the low vapor concentration of dry-conditioned bags. We therefore recommend conditioning entire batches of sample bags simultaneously with a moist, isotopically homogeneous atmosphere prior to each isotope sampling campaign.
Further, we emphasize that for any given vapor sample an isotopic shift must be expected between collection and subsequent lab-based analysis. This shift became apparent, e.g., during field application (Fig. 4) and the conditioning procedure (Fig. 6). It is characteristic and implicit to our method. However, it is proportional to the priming signal. Herewith, it becomes manageable through co-measuring calibration standards, prepared in identically pre-treated bags.
Our conditioning routine was performed manually, and its efficiency checked after every conditioning iteration. However, we are confident that it can be easily automated for future applications and its efficiency eventually assumed reliably without measurements as a matter of experience. Further, it seems that for the SD being a meaningful parameter for conditioning efficiency, the conditioning time steps need to match the projected sample storage time. This also calls for automating the conditioning procedure.
Our aim was to develop a low-cost solution as funding might not always be available, and we wanted to avoid the high per-unit costs of commercially available gas sampling bags. Moreover, diffusion-tight bags from, for example, Analyt-MTC are made from plastic-coated aluminum foil (Analyt-MTC Products, 2015), similar to the ones we used in our study. Therefore, similar adsorption issues must be expected due to the interior coating of the diffusive barrier. This means that before being readily available some kind of conditioning procedure would also have to be applied, even when reusing commercially available bags. Also, for any other sampling vessel a potential user will have to verify the suitability in a way similar to the one we describe: in terms of diffusion tightness, contamination, and adsorptive disturbances from the inner layer of the vessel material itself.
So far, we are satisfied with the outcome of our tests and the achievable precision and accuracy. Nonetheless, we are aware that our method is not yet ready to go, and further tests are needed before unknown field-collected samples can be processed. This does not just refer to the issue of potential temperature discrepancies between samples and standards. Moreover, resulting differences in vapor concentration must be considered, which likely induce additional challenges. The proportionality of the memory effect observed in this study, e.g., after applying the moist-air conditioning procedure, was likely because we were able to facilitate a consistent ratio of collected sample size and vapor reservoir absorbed to the bags' inner walls expressible, for example, in water vapor mole fractions. The effect of changes of this ratio has not been tested yet nor has a mathematical correction scheme accounting for this issue been found. Further, we are aware that the effort that needs to be put into assembling the necessary components as well as into pre-sample-collection tests and conditioning might still have a deterring effect on potential users of our method. The same probably holds for the costly alternatives available in the market for collecting gas samples into flexible containers that would at least dispense with the assembly efforts. In this context, we admit that the option to use inexpensive off-the-shelf components as employed by Magh et al. (2022) is certainly appealing. Also, the option to reliably reproduce vapor isotope values after 1 or more weeks of storage remains a worthwhile goal.
On the other hand, we believe that the otherness of our method also holds several advantages relative to previous approaches for the collection and analysis of discrete vapor samples. In contrast to Havranek et al. (2020, 2023), the non-automation allows for maximum temporal flexibility regarding experimental designs. The spatial distribution of sampling sites is not limited by connectivity to a central sampling system and associated tubing lengths. As all employed components are small and lightweight, they can easily be deployed even in terrains that are rather difficult to access. In contrast to Magh et al. (2022), the achieved precision and accuracy (Table 3) allow for the resolution of even fine-scaled environmental isotope variations. Practically, our method does not require any tools, not to mention specialty tools during handling or refurbishing of crimp-sealed glass bottles, and the lack of consumables such as septa makes it cost-efficient. The combination of the screw-lock mechanism and a sufficiently large sample volume is forgiving as it allows for multiple measurements from one sample bag. Also, flushing and refilling are possible without wearing of the closing mechanism. Using evacuated, inflatable containers renders any in-field flushing obsolete, thus reducing sampling time and increasing feasible temporal resolution. Further, it eliminates the risk of breaking glass during transport and handling. Finally, our method does not require additional gas sources during analysis and allows for directly interpretable measurement readings without the need to identify the relevant isotope data section via derivation of the vapor concentration readings.
We present a new method for the collection of discrete water vapor samples in the field and subsequent storage and isotope analysis in the lab. After systematic material testing (Appendix Table A1), we identified a combination of inflatable bag and closure type that guarantees airtightness and avoids sample contamination by material outgassing. Similar tests are also necessary for commercially available gas sampling bags. Our custom-made method uses off-the-shelf components only and is easy to use, cost-efficient, sustainable, and allows for multiple measurements. Further, it allows for direct interpretation of the obtained isotope results. The achieved precision and accuracy are not only suitable for labeling experiments but also sufficient for resolving natural variations of water stable isotope signatures. Preparation and co-measurement of calibration standards are indispensable for our approach in order to correct for the implicit shift of the obtained isotope signal, induced by the mandatory conditioning procedure. We are convinced that the conditioning procedure can be automated, which would further reduce the per-sample workload when reusing sampling bags. The presented approach allows for collecting vapor samples from soil matrix and plant tissues in remote settings without an isotope analyzer in the field. The method therefore widens the applicability of minimally invasive in situ approaches of matrix-bound water stable isotope observations. Hence, we are confident that our method will open new observatory paths and thus contribute to novel insights into hydrology, soil science, plant physiology, and related disciplines. Future tests will have to find ways of dealing with discrepancies in temperature, vapor concentration, or matrix potential among samples during collection and relative to co-prepared standards.
The procedure of testing and the requirements that had to be fulfilled in our study by the tested gas sampling bags are summarized in the protocol. Any other material or commercially available bags can also be evaluated by passing this protocol.
Table A1Suggested test protocol to identify suitable gas sampling bags for isotopic vapor.
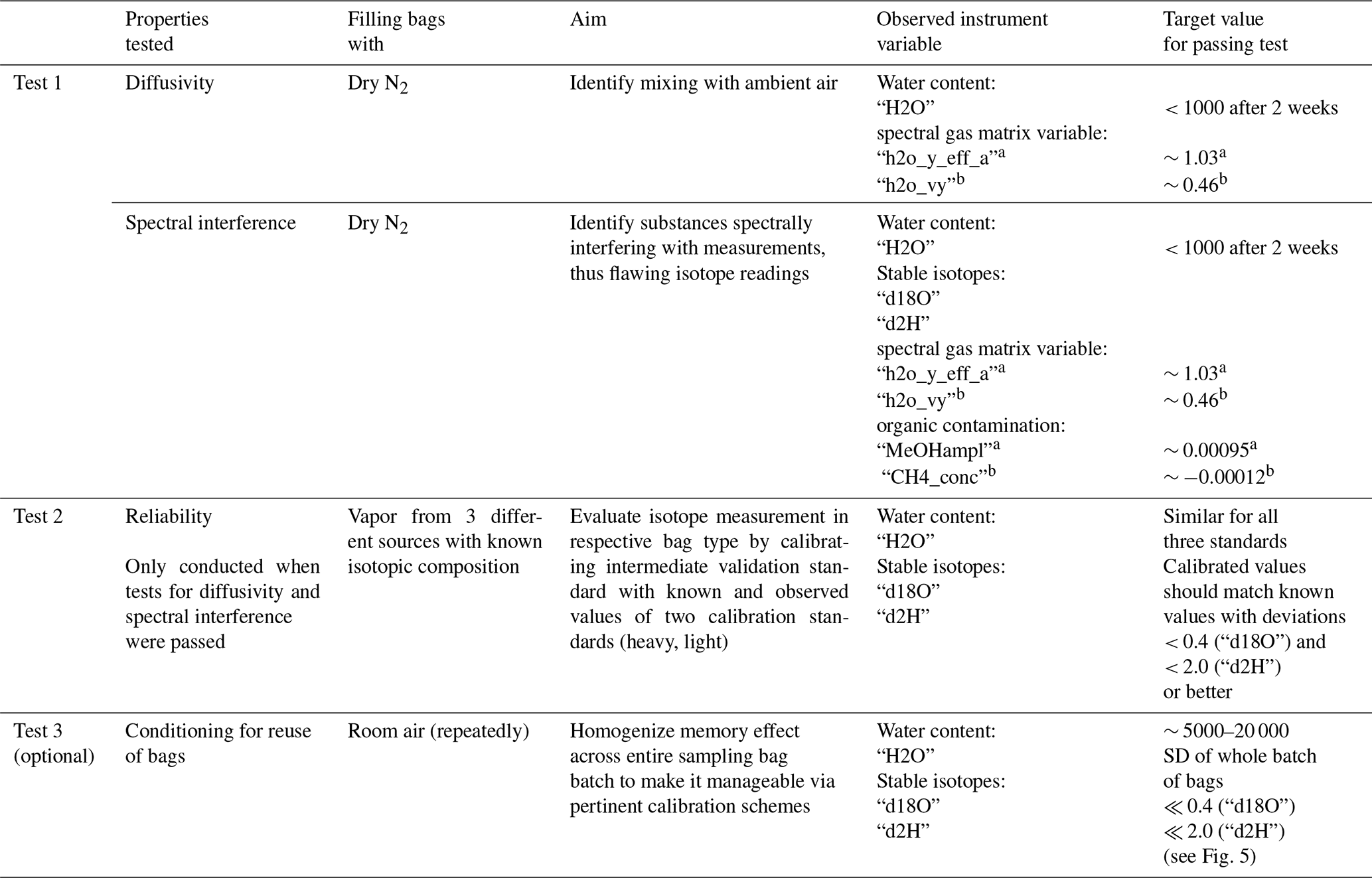
a L2120-i; b L2130-i.
In reused bags, we observed isotope data consistently trending towards the values recorded previously from the respective sample bags, although evacuated and flushed three times with dry N2. These memory effects were proportional to the differences between the current and the previous bag measurement (Fig. B1).

Figure B1Dual isotope plot of gasbag measurements of reused bags 5 d (open squares) and 10 d (open triangles) after all of them being filled with a constant flow rate of 150 mL min−1 via a WIP from one single isotopic reservoir (filled dot). The different colors indicate the differing isotopic levels of the previous samples, stored in the respective bags.
Data are available from the authors on request.
The supplement related to this article is available online at: https://doi.org/10.5194/hess-27-3701-2023-supplement.
BH, StS, MR, and MW designed the first part (material selection, field trial) of the experiments, BH and StS carried them out. BH and BG designed the second part (climate chamber, conditioning procedure) of the experiments and carried them out. BH, BG, and MW analyzed and interpreted the data. BH and BG prepared the manuscript with contributions from all co-authors.
At least one of the (co-)authors is a member of the editorial board of Hydrology and Earth System Sciences.
Publisher’s note: Copernicus Publications remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The authors thank Janine Heitzmann for her tireless commitment in the lab during material testing.
This research has been supported by the Deutsche Forschungsgemeinschaft (grant nos. GE 1090/10-1 and WE 4598/7-2) through the priority program SPP 1685 “Ecosystem Nutrition: Forest Strategies for limited Phosphorus Resources”.
This open-access publication was funded by the University of Freiburg.
This paper was edited by Thom Bogaard and reviewed by two anonymous referees.
Allison, G. B.: The relationship between 18O and deuterium in water in sand columns undergoing evaporation, J. Hydrol., 55, 163–169, https://doi.org/10.1016/0022-1694(82)90127-5, 1982.
Allison, G. B., Barnes, C. J., and Hughes, M. W.: The distribution of deuterium and 18O in dry soils 2. Experimental, J. Hydrol., 64, 377–397, https://doi.org/10.1016/0022-1694(83)90078-1, 1983.
Analyt-MTC Products: https://analyt-mtc.de/files/50/Produkte/4/Probenahme.pdf (last access: 20 July 2023), 2015 (in German).
Baer, D. S., Paul, J. B., Gupta, M., and O'Keefe, A.: Sensitive absorption measurements in the near-infrared region using off-axis integrated-cavity-output spectroscopy, Appl. Phys. B-Lasers O., 75, 261–265, https://doi.org/10.1007/s00340-002-0971-z, 2002.
Barnes, C. J. and Allison, G. B.: Tracing of water movement in the unsaturated zone using stable isotopes of hydrogen and oxygen, J. Hydrol., 100, 143–176, https://doi.org/10.1016/0022-1694(88)90184-9, 1988.
Berden, G., Peeters, R., and Meijer, G.: Cavity ring-down spectroscopy: experimental schemes and applications, Int. Rev. Phys. Chem., 19, 565–607, 2000.
Beyer, M., Koeniger, P., Gaj, M., Hamutoko, J. T., Wanke, H., and Himmelsbach, T.: A deuterium-based labeling technique for the investigation of rooting depths, water uptake dynamics and unsaturated zone water transport in semiarid environments, J. Hydrol., 533, 627–643, https://doi.org/10.1016/j.jhydrol.2015.12.037, 2016.
Brand, W. A., Geilmann, H., Crosson, E. R., and Rella, C. W.: Cavity ring-down spectroscopy versus high-temperature conversion isotope ratio mass spectrometry; a case study on δ2H and δ18O of pure water samples and alcohol/water mixtures, Rapid Commun. Mass Sp., 23, 1879–1884, 2009.
Brunel, J. P., Walker, G. R., Dighton, J. C., and Monteny, B.: Use of stable isotopes of water to determine the origin of water used by the vegetation and to partition evapotranspiration. A case study from HAPEX-Sahel, J. Hydrol., 188–189, 466–481, 1997.
Buttle, J. M. and Sami, K.: Recharge processes during snowmelt: An isotopic and hydrometric investigation, Hydrol. Process., 4, 343–360, https://doi.org/10.1002/hyp.3360040405, 1990.
Caldwell, M. M. and Richards, J. H.: Hydraulic lift: water efflux from upper roots improves effectiveness of water uptake by deep roots, Oecologia, 79, 1–5, 1989.
Craig, H.: Standard for reporting concentrations of deuterium and oxygen-18 in natural waters, Science, 133, 3467, 1833–1834, 1961a.
Craig, H.: Isotopic Variations in Meteoric Waters, Science, 133, 3465, https://doi.org/10.1126/science.133.3465.1702, 1961b.
Crosson, E. R.: A cavity ring-down analyzer for measuring atmospheric levels of methane, carbon dioxide, and water vapor, Appl. Phys. B-Lasers O., 92, 403–408, https://doi.org/10.1007/s00340-008-3135-y, 2008.
Darling, W. G. and Bath, A. H.: A stable isotope study of recharge processes in the English Chalk, J. Hydrol., 101, 31–46, https://doi.org/10.1016/0022-1694(88)90026-1, 1988.
Dawson, T. E. and Ehleringer, J. R.: Streamside trees that do not use stream water, Nature, 350, 335–337, 1991.
Dincer, T., Al-Mugrin, A., and Zimmermann, U.: Study of the infiltration and recharge through the sand dunes in arid zones with special reference to the stable isotopes and thermonuclear tritium, J. Hydrol., 23, 79–109, https://doi.org/10.1016/0022-1694(74)90025-0, 1974.
Drake, P. L. and Franks, P. J.: Water resource partitioning, stem xylem hydraulic properties, and plant water use strategies in a seasonally dry riparian tropical rainforest, Oecologia, 137, 321–329, 2003.
Dubbert, M., Cuntz, M., Piayda, A., Maguas, C., and Werner, C.: Partitioning evapotranspiration – testing the Craig and Gordon model with field measurements of oxygen isotope ratios of evaporative fluxes, J. Hydrol., 496, 142–153, 2013.
Dubbert, M., Cuntz, M., Piayda, A., and Werner, C.: Oxygen isotope signatures of transpired water vapor: the role of isotopic non-steady-state transpiration under natural conditions, New Phytol., 203, 1242–1252, 2014.
Ehleringer, J., Phillips, S., Schuster, W. F., and Sandquist, D.: Differential utilization of summer rains by desert plants, Oecologia, 88, 430–434, 1991.
Ehleringer, J. R., Roden, J., and Dawson, T. E.: Assessing ecosystem-level water relations through stable isotope ratio analyses, Methods in ecosystem science, edited by: Sala, O. E., Jackson, R., Mooney, H. A., and Howarth, R., Springer Verlag, New York, NY, USA, https://doi.org/10.1007/978-1-4612-1224-9_13, 181–198, 2000.
Fan, Y., Miguez-Macho, G., Jobbágy, E. G., Jackson, R. B., and Otero-Casal, C: Hydrologic regulation of plant rooting depth, P. Natl. Acad. Sci. USA, 114, 10572–10577, https://doi.org/10.1073/pnas.1712381114, 2017.
Foken, T.: Angewandte Meteorologie, Springer-Verlag, Berlin, Germany, 325 pp., https://doi.org/10.1007/978-3-642-25525-0_8, 2008.
Garvelmann, J., Külls, C., and Weiler, M.: A porewater-based stable isotope approach for the investigation of subsurface hydrological processes, Hydrol. Earth Syst. Sci., 16, 631–640, https://doi.org/10.5194/hess-16-631-2012, 2012.
Gazis, C. and Feng, X.: A stable isotope study of soil water: evidence for mixing and preferential flow paths, Geoderma, 119, 97–111, https://doi.org/10.1016/s0016-7061(03)00243-x, 2004.
Gessler, A., Bächli, L., Rouholahnejad-Freund, E., Treydte, K., Schaub, M., Haeni, M., Weiler, M., Seeger, S., Marshall, J., Hug, C., Zweifel, R., Hagedorn, F., Rigling, A., Saurer, M., and Meusburger, K.: Drought reduces water uptake in beech from the drying topsoil, but no compensatory uptake occurs from deeper soil layers, New Phytol., 233, 194–206, https://doi.org/10.1111/nph.17767, 2022.
Gralher, B., Herbstritt, B., Weiler, M., Wassenaar, L. I., and Stumpp, C.: Correcting Laser-Based Water Stable Isotope Readings Biased by Carrier Gas Changes, Environ. Sci. Technol., 50, 7074–7081 https://doi.org/10.1021/acs.est.6b01124, 2016.
Gralher, B., Herbstritt, B., and Weiler, M.: Technical note: Unresolved aspects of the direct vapor equilibration method for stable isotope analysis (δ18O, δ2H) of matrix-bound water: unifying protocols through empirical and mathematical scrutiny, Hydrol. Earth Syst. Sci., 25, 5219–5235, https://doi.org/10.5194/hess-25-5219-2021, 2021.
Gupta, P., Noone, D., Galewsky, J., Sweeney, C. and Vaughn, B. H.: Demonstration of high-precision continuous measurements of water vapor isotopologues in laboratory and remote field deployments using wavelength-scanned cavity ring-down spectroscopy (WS-CRDS) technology, Rapid Commun. Mass. Sp., 23, 2534–2542, 2009.
Havranek, R. E., Snell, K. E., Davidheiser-Kroll, B, Bowen, G. J., and Vaughn, B.: The Soil Water Isotope Storage System (SWISS): An integrated soil water vapor sampling and multiport storage system for stable isotope geochemistry, Rapid Commun. Mass. Sp., 34, e8783, https://doi.org/10.1002/rcm.8783, 2020.
Havranek, R. E., Snell, K., Kopf, S., Davidheiser-Kroll, B., Morris, V., and Vaughn, B.: Technical note: Lessons from and best practices for the deployment of the Soil Water Isotope Storage System, Hydrol. Earth Syst. Sci., 27, 2951–2971, https://doi.org/10.5194/hess-27-2951-2023, 2023.
Hendry, M. J., Richman, B., and Wassenaar, L. I.: Correcting for Methane Interferences on δ2H and δ18O Measurements in Pore Water Using H2O(liquid)-H2O(vapor) Equilibration Laser Spectroscopy, Anal. Chem., 83, 5789–5796, 2011.
Hendry, M. J., Schmeling, E., Wassenaar, L. I., Barbour, S. L., and Pratt, D.: Determining the stable isotope composition of pore water from saturated and unsaturated zone core: improvements to the direct vapour equilibration laser spectrometry method, Hydrol. Earth Syst. Sci., 19, 4427–4440, https://doi.org/10.5194/hess-19-4427-2015, 2015.
Herbstritt, B., Gralher, B., and Weiler, M.: Continuous in situ measurements of stable isotopes in liquid water, Water Resour. Res., 48, W03601, https://doi.org/10.1029/2011WR011369, 2012.
Herbstritt, B., Gralher, B., and Weiler, M.: Continuous, near-real-time observations of water stable isotope ratios during rainfall and throughfall events, Hydrol. Earth Syst. Sci., 23, 3007–3019, https://doi.org/10.5194/hess-23-3007-2019, 2019.
Horita, J. and Kendall, C.: Stable isotope analysis of water and aqueous solutions by conventional dual-inlet mass spectrometry, in: Handbook of stable isotope analytical techniques, edited by: de Groot, P. A., Elsevier, Amsterdam, the Netherlands, 1–37, https://doi.org/10.1016/B978-044451114-0/50003-X, 2004.
Hsieh, J. C. C., Chadwick, O. A., Kelly, E. F., and Savin, S. M.: Oxygen isotopic composition of soil water: Quantifying evaporation and transpiration, Geoderma, 82, 269–293, https://doi.org/10.1016/s0016-7061(97)00105-5, 1998.
Kerstel, E. and Gianfrani, L.: Advances in laser-based isotope ratio measurements: selected applications, Appl. Phys. B-Lasers O., 92, 439-449, 2008.
Koeniger, P., Gaj, M., Beyer, M., and Himmelsbach, T.: Review on soil water isotope-based groundwater recharge estimations, Hydrol. Process., 30, 2817–2834, https://doi.org/10.1002/hyp.10775, 2016.
Kühnhammer, K., Dahlmann, A., Iraheta, A., Gerchow, M., Birkel, C., Marshall, J. D., and Beyer, M.: Continuous in situ measurements of water stable isotopes in soils, tree trunk and root xylem: Field approval, Rapid Commun. Mass Sp., 36, e9232, https://doi.org/10.1002/rcm.9232, 2022.
Laudon, H., Seibert, J., Köhler, S., and Bishop, K.: Hydrological flow paths during snowmelt: Congruence between hydrometric measurements and oxygen 18 in meltwater, soil water, and runoff, Water Resour. Res., 40, W03102, https://doi.org/10.1029/2003wr002455, 2004.
Magh, R.-K., Gralher, B., Herbstritt, B., Kübert, A., Lim, H., Lundmark, T., and Marshall, J.: Technical note: Conservative storage of water vapour – practical in situ sampling of stable isotopes in tree stems, Hydrol. Earth Syst. Sci., 26, 3573–3587, https://doi.org/10.5194/hess-26-3573-2022, 2022
Majoube, M.: Fractionnement en oxygen et en deuterium entre l'eau et sa vapeur, J. Chim. Phys., 68, 1423–1436, 1971.
Marshall, J. D., Cuntz, M., Beyer, M., Dubbert, M., and Kuehnhammer, K.: Borehole Equilibration: Testing a New Method to Monitor the Isotopic Composition of Tree Xylem Water in situ, Front. Plant Sci., 11, 358, https://doi.org/10.3389/fpls.2020.00358, 2020.
McDonnell, J. J.: A Rationale for Old Water Discharge Through Macropores in a Steep, Humid Catchment, Water Resour. Res., 26, 2821–2832, https://doi.org/10.1029/WR026i011p02821, 1990.
Meißner, M., Köhler, M., Schwendenmann, L., and Hölscher, D.: Partitioning of soil water among canopy trees during a soil desiccation period in a temperate mixed forest, Biogeosciences, 9, 3465–3474, https://doi.org/10.5194/bg-9-3465-2012, 2012.
Meunier, F., Rothfuss, Y., Bariac, T., Biron, P., Richard, P., Durand, J., Couvreur, V., Vanderborght, J., and Javaux, M.: Measuring and Modeling Hydraulic Lift of Lolium multiflorum Using Stable Water Isotopes, Vadose Zone J., 17, 160134, https://doi.org/10.2136/vzj2016.12.0134, 2018.
Munksgaard, N. C., Wurster, C. M., and Bird, M. I.: Continuous analysis of d18O and δD values of water by diffusion sampling cavity ring-down spectrometry: a novel sampling device for unattended field monitoring of precipitation, ground and surface waters, Rapid Commun. Mass Sp., 25, 3706–3712, https://doi.org/10.1002/rcm.5282, 2011.
Picarro Document Library: L2130-i Analyzer Datasheet, https://www.picarro.com/support/library/documents/l2130i_analyzer_datasheet# (last access: 20 July 2023), 2021.
Quade, M., Klosterhalfen, A., Graf, A., Brüggemann, N., Hermes, N., Vereecken, H., and Rothfuss, Y.: In-situ monitoring of soil water isotopic composition for partitioning of evapotranspiration during one growing season of sugar beet (Beta vulgaris), Agr. Forest Meteorol., 266–267, 53–64, https://doi.org/10.1016/j.agrformet.2018.12.002, 2019.
Rothfuss, Y. and Javaux, M.: Reviews and syntheses: Isotopic approaches to quantify root water uptake: a review and comparison of methods, Biogeosciences, 14, 2199–2224, https://doi.org/10.5194/bg-14-2199-2017, 2017.
Rothfuss, Y., Biron, P., Braud, I., Canale, L., Durand, J. L., Gaudet, J. P., Richard, P., Vauclin, M., and Bariac, T.: Partitioning evapotranspiration fluxes into soil evaporation and plant transpiration using water stable isotopes under controlled conditions, Hydrol. Process., 24, 3177–3194, https://doi.org/10.1002/Hyp.7743, 2010.
Rothfuss, Y., Vereecken, H., and Brüggemann, N.: Monitoring water stable isotopic composition in soils using gas-permeable tubing and infrared laser absorption spectroscopy, Water Resour. Res., 49, 3747–3755, https://doi.org/10.1002/wrcr.20311, 2013.
Saxena, R.: Seasonal variations of oxygen-18 in soil moisture and estimation of recharge in Esker and Moraine formations, paper presented at the Nordic Hydrological Conference (Nyborg, Denmark, August, 1984), Hydrol. Res., 15, 235–242, https://doi.org/10.2166/nh.1984.0020, 1984.
Schwinning, S., Davis, K., Richardson, L., and Ehleringer, J. R.: Deuterium enriched irrigation indicates different forms of rain use in shrub/grass species of the Colorado Plateau, Oecologia, 130, 345–355, 2002.
Seeger, S. and Weiler, M.: Temporal dynamics of tree xylem water isotopes: in situ monitoring and modeling, Biogeosciences, 18, 4603–4627, https://doi.org/10.5194/bg-18-4603-2021, 2021.
Sklash, M. G. and Farvolden, R. N.: The role of groundwater in storm runoff, J. Hydrol., 43, 45–65, https://doi.org/10.1016/0022-1694(79)90164-1, 1979.
Stewart, M. K. and McDonnell, J. J.: Modeling base flow soil water residence times from deuterium concentrations, Water Resour. Res., 27, 2681–2693, https://doi.org/10.1029/91wr01569, 1991.
Volkmann, T. H. M. and Weiler, M.: Continual in situ monitoring of pore water stable isotopes in the subsurface, Hydrol. Earth Syst. Sci., 18, 1819–1833, https://doi.org/10.5194/hess-18-1819-2014, 2014.
Volkmann, T. H. M., Kühnhammer, K., Herbstritt, B., Gessler, A., and Weiler, M.: A method for in situ monitoring of the isotope composition of tree xylem water using laser spectroscopy, Plant Cell Environ., 39, 2055–2063, https://doi.org/10.1111/pce.12725, 2016a.
Volkmann, T. H. M., Haberer, K., Gessler, A., and Weiler, M.: High-resolution isotope measurements resolve rapid ecohydrological dynamics at the soil-plant interface, New Phytol., 210, 839–849, https://doi.org/10.1111/nph.13868, 2016b.
Walker, G. R., Hughes, M. W., Allison, G. B., and Barnes, C. J.: The movement of isotopes of water during evaporation from a bare soil surface, J. Hydrol., 97, 181–197, https://doi.org/10.1016/0022-1694(88)90114-x, 1988.
Wang, X. F. and Yakir, D.: Using stable isotopes of water in evapotranspiration studies, Hydrol. Process., 14, 1407– 1421, https://doi.org/10.1002/1099-1085(20000615)14:8<1407::AID-HYP992>3.0.CO;2-K, 2000.
Wang, L., Good, S. P., Caylor, K. K., and Cernusak, L. A.: Direct quantification of leaf transpiration isotopic composition, Agr. Forest Meteorol., 154–155, 127–135, 2012.
Wassenaar, L., Hendry, M., Chostner, V., and Lis, G.: High Resolution Pore Water δ2H and δ18O Measurements by H2O(liquid)-H2O(vapor) Equilibration Laser Spectroscopy, Environ. Sci. Technol., 42, 9262–9267, 2008.
Werner, R. A. and Brand, W. A.: Referencing strategies and techniques in stable isotope ratio analysis, Rapid Commun Mass Sp., 15, 501–519, https://doi.org/10.1002/rcm.258, 2001.
West, A. G., Patrickson, S. J., and Ehleringer, J. R.: Water extraction times for plant and soil materials used in stable isotope analysis, Rapid Commun. Mass Sp., 20, 1317–1321, 2006.
West, A. G., Goldsmith, G. R., Brooks, P. D., and Dawson, T. E.: Discrepancies between isotope ratio infrared spectroscopy and isotope ratio mass spectrometry for the stable isotope analysis of plant and soil waters, Rapid Commun. Mass Sp., 24, 1948–1954, 2010.
Yepez, E. A., Huxman, T. E., Ignace, D. D., English, N. B., Weltzin, J. F., Castellanos, A. E., and Williams, D. G.: Dynamics of transpiration and evaporation following a moisture pulse in semiarid grassland: A chamber-based isotope method for partitioning flux components, Agr. Forest Meteorol., 132, 359–376, https://doi.org/10.1016/j.agrformet.2005.09.006, 2005.
Zimmermann, U., Ehhalt, D., and Münnich, K. O.: Soil water movement and evapotranspiration: changes in the isotopic composition of the water, Isotopes in Hydrology, IAEA, Vienna, Austria, 567–585, ISSN 0074-1884, 1967.
- Abstract
- Introduction
- Methodology
- Results
- Discussion
- Conclusions
- Appendix A: Protocol for identifying appropriate sampling bags
- Appendix B: Memory effects in reused bags
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement
- Abstract
- Introduction
- Methodology
- Results
- Discussion
- Conclusions
- Appendix A: Protocol for identifying appropriate sampling bags
- Appendix B: Memory effects in reused bags
- Data availability
- Author contributions
- Competing interests
- Disclaimer
- Acknowledgements
- Financial support
- Review statement
- References
- Supplement